Dexamethasone Interferes with Autophagy and Affects Cell Survival in Irradiated Malignant Glioma Cells
Article information

Abstract
Objective
Radiation is known to induce autophagy in malignant glioma cells whether it is cytocidal or cytoprotective. Dexamethasone is frequently used to reduce tumor-associated brain edema, especially during radiation therapy. The purpose of the study was to determine whether and how dexamethasone affects autophagy in irradiated malignant glioma cells and to identify possible intervening molecular pathways.
Methods
We prepared p53 mutant U373 and LN229 glioma cell lines, which varied by phosphatase and tensin homolog (PTEN) mutational status and were used to make U373 stable transfected cells expressing GFP-LC3 protein. After performing cell survival assay after irradiation, the IC50 radiation dose was determined. Dexamethasone dose (10 μM) was determined from the literature and added to the glioma cells 24 hours before the irradiation. The effect of adding dexamethasone was evaluated by cell survival assay or clonogenic assay and cell cycle analysis. Measurement of autophagy was visualized by western blot of LC3-I/LC3-II and quantified by the GFP-LC3 punctuated pattern under fluorescence microscopy and acridine orange staining for acidic vesicle organelles by flow cytometry.
Results
Dexamethasone increased cell survival in both U373 and LN229 cells after irradiation. It interfered with autophagy after irradiation differently depending on the PTEN mutational status : the autophagy decreased in U373 (PTEN-mutated) cells but increased in LN229 (PTEN wild-type) cells. Inhibition of protein kinase B (AKT) phosphorylation after irradiation by LY294002 reversed the dexamethasone-induced decrease of autophagy and cell death in U373 cells but provoked no effect on both autophagy and cell survival in LN229 cells. After ATG5 knockdown, radiation-induced autophagy decreased and the effect of dexamethasone also diminished in both cell lines. The diminished autophagy resulted in a partial reversal of dexamethasone protection from cell death after irradiation in U373 cells; however, no significant change was observed in surviving fraction LN229 cells.
Conclusion
Dexamethasone increased cell survival in p53 mutated malignant glioma cells and increased autophagy in PTEN-mutant malignant glioma cell but not in PTEN-wildtype cell. The difference of autophagy response could be mediated though the phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin signaling pathway.
INTRODUCTION
Glioblastoma multiforme (GBM), an aggressive primary brain tumor, is characterized by invasive tumor growth and remote extension within the brain at the later course of disease. Thus, surgery plays a limited role in the treatment of this tumor, and a cure remains elusive. Currently, the standard therapy for GBM is surgical removal, followed by concomitant chemoradiotherapy and adjuvant chemotherapy with temozolomide [10].
Radiotherapy is a major cancer treatment modality for solid tumors as it kills cancer cells and shrinks the tumor either before or after surgical resection [4]. However, two limitations to treatment success are the resistance of glioma cells to radiation and dose-related adverse events, resulting in poor prognosis.
Dexamethasone, a glucocorticoid steroid, has been used for decades in the treatment of malignant gliomas to reduce tumor-associated cerebral edema, especially during radiation therapy. However, the effect of dexamethasone on cancer cell growth and patient survival remains contentious. Many studies have suggested that it may protect cancer cells from cell death induced by chemotherapy/radiotherapy, thus promoting cancer cell survival [6,9,28,30].
Autophagy is a vital catabolic mechanism where cells digest and recycle their own organelles for maintaining cellular homeostasis [13,29]. Glucocorticoids are known to induce catabolism as they frequently cause steroid-induced diabetes mellitus. In this context, dexamethasone is also known to induce autophagy in many cancer cells or immortalized cell lines in vitro [23,35,37,43]. Researchers have suggested that autophagy could be important in determining the response of tumor cells to anticancer therapy [15,24]. Ionizing radiation-induced autophagy in many solid tumors, including glioma, may trigger tumor cell survival or cell death [8,27,40,41]. Cross-talk between apoptosis and autophagy has been suggested but remains unclear [18,24]. In a previous study, we demonstrated that autophagy is partly responsible for radiation-induced cell death or apoptosis in malignant glioma cells [17].
Phosphatase and tensin homolog (PTEN) is a tumor suppressor gene that is frequently mutated in GBM. It encodes a dual specificity phosphatase that negatively regulates the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway, which is a key regulator of autophagy [31]. The PI3K/AKT pathway plays an important role in cancer development, and its deregulation via the loss of PTEN function is a common occurrence in many aggressive cancers such as malignant gliomas [7,22,31]. In addition, AKT promotes cell survival by inhibiting apoptosis through the phosphorylation and inactivation of several targets after exposure to chemotherapeutic agents or radiation [7,25,39]. Thus, it plays an important role in radiation-induced autophagy in glioma cells.
The purpose of this study was to determine whether dexamethasone affects autophagy and cell survival in irradiated malignant glioma cells and by what mechanism. The effect of dexamethasone on autophagy has never been evaluated in irradiated malignant glioma cells. We used two p53 mutant glioma cell lines that varied by PTEN mutational status and assessed the effect of dexamethasone on cell death and autophagy.
Materials and methods
The Institutional Review Board of National Cancer Center exempted any registration or permission for in vitro cell line experiment and publication of its result unless it uses animal model or patient’s derived tissue. National Cancer Center exempted any informed consent for in vitro study including this study.
Cell culture and irradiation
Human glioblastoma cell lines, LN229 and U373, were obtained from the American Type Culture Collection. Cells were grown in Dulbecco’s modified Eagle medium containing 10% fetal bovine serum and 1% penicillin/streptomycin at 37℃, 5% CO2. Cells were treated with 10 μM dexamethasone sodium at the time of plating. At 24 hours after plating, they were irradiated with a Gammacell 1000 Elite Cesium137 source (MDS Nordion, Ottawa, Canada) for a calculated dose by dose rate of 0.0416 Gy/second.
RNA interference
To suppress autophagosome formation, we knocked down the autophagy-related 5 (ATG5) protein in both cell lines. Knockdown of ATG5 expression was done using small interfering RNA (SiRNA) and a negative control bought from Ambion (Life Technologies, Calsbad, CA, USA) transfected into the glioma cells using the Lipofectamine 2000 reagent (Invitrogen, Calsbad, CA, USA) in accordance with the manufacturer’s instructions. After 24 hours, cells were used in the experiment.
Cell survival assay
Two methods of cell viability were employed : cell-counting using an automated cell counter (NanoEnTek, Seoul, Korea) after trypan blue staining and the clonogenic assay. During the survival assay, cells were plated on 60-mm culture plates (2×104 cells). After 24 hours, 10 μM dexamethasone was added, and 24 hours later, the cells were subjected to different doses of radiation : 0, 2, 5, 10, 15, and 20 Gy. Cells were then incubated at 37°C with regular media changes for 8–10 days until the control was 100% confluent. Cells were washed twice with phosphate-buffered saline (PBS), trypsinized with 1 mL trypsin-EDTA, stained with trypan blue, and counted using an automated cell counter. The percentage of viable cells was calculated from the ratio of treated cells to normal control cells. Three independent experiments were carried out, and the mean value was determined.
For the colony-forming assay, cells were plated at clonogenic density (1.0×103) in a 60-mm culture plate, then treated with dexamethasone (10 μM) and a radiation dose (10 Gy) as per the experiment setup. Cells were incubated for 8–10 days with regular media changes after treatment. Cells were fixed with 4% paraformaldehyde for 20 minutes, then stained with 0.4% crystal violet for 20 minutes, washed under running distilled water, and allowed to dry for 2 hours. A cluster of 50 cells or more was scored as a colony.
Cell cycle analysis
Cells were plated on 60-mm plates (1×105 cells) and pretreated with 10 μM dexamethasone; 24 hours later, they were subjected to 10 Gy radiation during the log growth phase. At 72 hours after treatment, cells (including non-adherent cells) were collected, centrifuged twice at 1500 rpm for 5 minutes in PBS to remove trypsin, then fixed slowly by adding drops of 70% cold ethanol and kept overnight at 4°C.
The fixed cells were washed twice with cold PBS at 2000 rpm for 10 minutes each. Next, these cells were suspended in 500 μL PI (50 μg/mL in PBS) containing 100 μg/mL RNase A (CosmoGenetech, Seoul, Korea) and then incubated in darkness at room temperature for 30 minutes. DNA content was analyzed using a fluorescence-activated cell sorter (BD LSRFortessa; BD Biosciences, Franklin Lakes, NJ, USA) and the FACSDiva software (BD Biosciences).
Quantitative measurement of autophagy using acridine orange
For acidic vesicular organelle (AVO) staining, pretreated cells (as described above) were incubated at 37°C for 30 minutes in a pre-warmed culture media containing acridine orange (1 μg/mL). After trypsinization and washing twice in PBS, pelleted cells were suspended in PBS and kept on ice, protected from light with an aluminum foil cover, and subjected to a fluorescence-activated cell sorter (BD LSRfortessa; BD Biosciences) and data analysis by BD FACSDiva software (BD Biosciences).
Autophagy measurement using GFP-LC3
Cells were transfected with a GFP-LC3 expression plasmid incorporated into the lentiviral vector using the Lipofectamine 2000 reagent (Invitrogen). Selection was made with puromycin to establish stably transfected cells expressing GFP-LC3 fluorescence, which was confirmed microscopically before the experiment. Three days following 10 Gy irradiation (pretreatment with 10 μM dexamethasone as described above), cells were observed under a fluorescence microscope for GFP-LC3 f luorescence, and LC3-punctuated cells were counted. The average percentage of autophagic cells was calculated from the ratio of GFP-LC3-punctuated cells to normal cells only bearing GFP-LC3 fluorescence per high power field (×200).
Confocal fluorescence microscope
GFP-LC3–transfected cells (1×105) were plated on a glass coverslip coated with poly-D-lysine in a 60-mm culture plate for 24 hours, then pretreated with dexamethasone. At 24 hours after pretreatment, cells were irradiated using a single 10 Gy dose, then incubated at 37°C for 3 days before analysis. Cells were fixed in 4% paraformaldehyde with DAPI staining for 30 minutes at room temperature. Fixed cells were washed with Dulbecco’s PBS and mounted on a glass slide using mounting medium. The glass slide with a coverslip was inverted on a confocal microscope mounting. Photographs were taken of cells expressing GFP-LC3 protein and punctuated spots (autophagosome) were analyzed with ZEISS software (Zeiss, Gena, Germany).
Western blot analysis
Cells were cultured on a 60-mm plate (1×105 cells), treated and incubated for the required time (as described above), washed and trypsinized, and then centrifuged to form cell pellets. These pellets were lysed in a RIPA lysis buffer with proteases and phosphatase inhibitor cocktail (100X, GenDEPOT, Katy, TX, USA) for at least 30 minutes on ice, then cleared by centrifugation for 20 minutes at 12000 rpm and 4°C. The concentration of protein was determined using bovine serum albumin protein standard and measured with spectrophotometry at 562 nm. The protein was heated at 95°C for 5 minutes and resolved by 12% and 15% polyacrylamide gel electrophoresis using mini-PROTEAN; it ran at 80 V for the first 30 minutes and later 125 V for 1 hour. The protein was transferred onto a nitrocellulose membrane at 250 A for 90 minutes at 4°C and then blocked for 1 hour with nonfat dry milk in 0.1% Tween in tris-buffered saline. The membrane was incubated at 4°C overnight in different primary antibodies : anti-LC3, anti-AKT, pAKT, anti-ATG5 (Novus Biologicals, Littleton, CO, USA), and anti-β-actin (Sigma-Aldrich, St. Louis, MO, USA). This membrane was then washed three times in TBST for 10 minutes and incubated with horseradish peroxidase-conjugated secondary antibody for 1 hour at room temperature. The protein complex was detected using western blot detection kit (Fisher Scientific, Waltham, MA, USA) A and B mixed in the ratio of 1 : 1 and visualized using an enhanced chemiluminescence developed on an X-ray film.
Statistical analysis
Three independent experiments were carried out (triplicate), and results were expressed as the mean±standard error of the mean. Statistical significance was calculated with a two-tailed unpaired Student t-test using GraphPad Prizm (ver. 6.0; GraphPad software, La Jolla, CA, USA). A p-value less than 0.05 was considered to be statistically significant.
RESULTS
Dexamethasone protects glioma cells from cell death after irradiation
We used a dexamethasone dose of 10 μM, which was determined by the literature as the highest clinically achievable plasma concentration [21,33], and applied a pre-determined radiation dose ranging from 2 Gy to 20 Gy. Dexamethasone alone did not affect cell proliferation at 10 μM (Supplementary Fig. 1). Cell survival after irradiation was compared to evaluate the effect of pretreating with dexamethasone (Fig. 1). The addition of dexamethasone increased the cell survival significantly in both U373 (p=0.02) and LN229 (p=0.03) cells. Based on these results, 10 Gy was used as the approximated inhibitory dose 50 (IC50) for further experiments to observe apoptosis and autophagy as calculated IC50 values were from 8.92 to 9.52 Gy from the assumption of sigmoid dose-response.

Dex increases survival in both U373 and LN229 glioma cell lines after irradiation. Malignant glioma cells were pretreated with 10 μM Dex and different doses of irradiation. both U373 (A, p=0.02) and LN229 (b, p=0.03) showed significant increases in survival. RT : radiation, Dex : dexamethasone.
Cell cycle analysis after irradiation with or without dexamethasone
The steroid effect on cell cycles was observed 3 days after irradiation using flow cytometry analysis (Fig. 2). After 10 Gy irradiation, the G2/M phase accumulation was significantly increased in radiation+dexamethasone (RT+Dex) compared to radiation only (RT) in both U373 (48% vs. 42%) and LN229 (44% vs. 32%) cell lines (p=0.021 and 0.017, respectively). Meanwhile, subG1 accumulation was significantly decreased with RT+Dex compared to RT in both U373 (9.8% vs. 19%, p=0.019) and LN229 cells (8.2% vs. 20%, p=0.02, respectively). These data together suggest that G2/M arrest and subG1 accumulation of a cell population at a lethal radiation dose is significantly interrupted by dexamethasone.

Dex interferes with the cell cycle change after irradiation in both U373 and LN229 cells. A : Flow cytometry at 3 days after irradiation (10 Gy) with or without Dex. b and C : Proportional analysis of cell cycle fraction revealed radiation-induced G2/M arrest and subG1 accumulation were interrupted significantly by the addition of Dex in both U373 and LN 229. *p<0.05. PI : propidium iodide, RT : radiation, Dex : dexamethasone.
Dexamethasone interferes autophagy after irradiation differently according to PTEN status
Because autophagy was a primary response to irradiation of malignant glioma cell lines in our previous study [17], we measured autophagy quantitatively to evaluate the inf luence of dexamethasone after irradiation. After acridine orange staining of AVOs, we subjected cells to flow cytometry analysis to measure autophagy (Fig. 3). For U373 cells, the AVO-containing cells decreased significantly in RT+Dex compared with RT (41% vs. 52%, p=0.01). For LN229 cells, AVO-containing cells increased as much as 56% in RT. These cells significantly increased further in RT+Dex (73%, p=0.01).

Measurement of acidic vesicular organelles (AVOs) as an indicator of autophagy after irradiation (10 Gy). A and C : Quantitative analysis of AVO revealed that dexamethasone (b) decreases autophagy in U373 cells (p=0.010) but (D) increases autophagy in LN229 cells (p=0.010). *p<0.05. RT : radiation, Dex : dexamethasone, FITC-A : fluorescein isothiocyanate.
The use of western blot analysis to evaluate the conversion of LC3-I to LC3-II conjugate is a standard indicator of autophagy [32]. In U373 cell lines, drastic reduction in the LC3-II band was found in RT+Dex compared with RT cells. By contrast, in LN229 cell lines, a significant increase of LC3-II was found in RT+Dex compared RT (Fig. 4A). These results are in accordance with quantitative measurement of AVO-indicating autophagy.
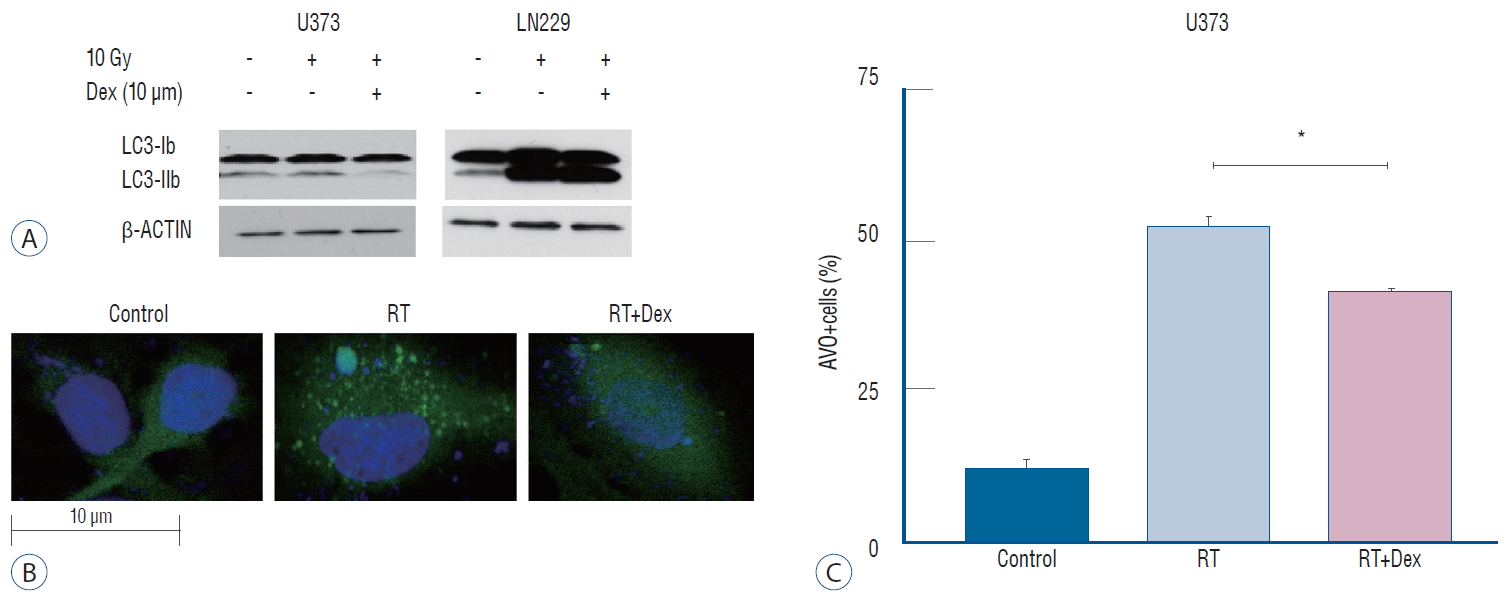
Illustration of Dex’s effect on radiation-induced autophagy in glioma cells. A : Dex inhibits autophagosome formation in U373 cells but promotes LC3-II conjugation in LN 229 cells on western blot. b : Fluorescence confocal microscope pictures (×2000) showing GFP-LC3 punctuated cells after irradiation (middle), which decreased with Dex (right) in U373 cells. C : Graphs indicating reduced number of GFP-LC3 punctuated cells with Dex (p=0.016) in U373 cells. Three independent experiments were carried out and the mean value is presented. *p<0.05. RT : radiation, Dex : dexamethasone, AVO : acidic vesicular organelle.
We further confirmed the inhibition of autophagy after irradiation by dexamethasone using GFP-LC3 stably transfected U373 cells (Fig. 4B). The expression level of GFP-LC3-punctuated cells showed autophagy increased after irradiation. Cells pretreated with dexamethasone (RT+Dex) exhibited less GTP-LC3 granules than those treated by RT (Fig. 4C; 30% vs. 15%, p=0.016).
Taken together, the data suggest that dexamethasone interferes with autophagy after irradiation, and the inf luence is different depending on the PTEN mutational status of the glioma cell.
Dexamethasone-induced reduced cell death is reversed with AKT inhibitor in PTEN non-functional U373 cells after irradiation
As PI3K/AKT pathway activation is the alleged cellular response to radiation-induced damage in some glioma cells [19], we confirmed phosphorylation of AKT in response to radiation in PTEN-mt U373 cells but not in PTEN-wt LN229 cells (Supplementary Fig. 2). Then, we performed a colony-forming assay to determine the effect of dexamethasone on cell survival after irradiation. When dexamethasone was added to the irradiated malignant glioma cells (RT+Dex), relative cell survival fractions were significantly increased compared with RT in both U373 and LN229 cells (Fig. 5).

Colony-forming assay after Dex and radiation exposure with or without phospho-AKT inhibitor (LY294002). A : Addition of Dex and LY294002 results in the reversal of decreased cell death with Dex in U373 cells (p=0.0052). b : It provoked no significant change (p>0.05) in LN229. The experiment was in triplicate and the mean value was presented. C : Photography of colony-forming assay plate of LN229 cells. *p<0.05. RT : radiation therapy, Dex : dexamethasone, LY : LY294002.
Next, we used the AKT inhibitor LY294002 to evaluate whether the dexamethasone effect on radiation-induced cell death is affected by the interruption of AKT activation according to PTEN status. The addition of the AKT inhibitor (RT+Dex+Ly) significantly reversed the dexamethasone-induced increased colony-forming ability in U373 cells (Fig. 5A; 0.45 vs. 0.34, p=0.0052). However, the addition of LY294002 provoked no significant change in colony number in LN229 cells pretreated with dexamethasone (p>0.05, Fig. 5B). These data demonstrate that the dexamethasone effect on irradiated glioma cells is mediated via AKT activation in PTEN-mt glioma cells, even though autophagy activity regulated by dexamethasone treatment differs by cell line.
Phospho-AKT inhibition interferes with the dexamethasone effect on autophagy formation after irradiation in U373 glioma cells
Next, we tested whether the effect of the AKT inhibitor on the dexamethasone-induced reduction of cell death after irradiation in U373 cells is related to autophagy activity (Fig. 6). Quantitative measurement of autophagy by AVO counting revealed that autophagy activity was significantly recovered by AKT inhibition (RT+Dex+Ly) when irradiated PTEN-mt U373 cells were pretreated with dexamethasone (RT+Dex) (74% vs. 50%, p=0.038); no discernible change was observed in PTEN-wt LN229 cells. This increased autophagy by an AKT inhibitor in U373 cells appeared to reverse the autophagy inactivation induced by dexamethasone after irradiation. Altogether, these data confirm that dexamethasone may be partly acting through the PI3K/AKT/mTOR pathway in PTEN non-functional glioma cells.

Phospho-AKT inhibition affects autophagy following Dex interference in irradiated malignant glioma cells. A : In U373, AVO expression increases significantly (RT+Dex+Ly vs. RT+Dex, p=0.038), indicating increased autophagy. b : In LN229, no significant change of autophagy is seen after blockage of p-AKT. *p<0.05. AVO : acidic vesicular organelle, RT : radiation therapy, Dex : dexamethasone, LY : LY294002.
Autophagy inhibition diminishes dexamethasone-induced increased colony-forming ability after irradiation not in PETN-mutant U373 cells
To determine how the knockdown of autophagy-initiating protein affect the irradiate malignant glioma cells, we knock down ATG5 protein with siRNA. After confirming ATG5 knockdown by western blot (Supplementary Fig. 3A), we measured the autophagy activity after irradiation by western blot of LC3 protein (Supplementary Fig. 3B). As expected, knock down of ATG5 significantly decreases LC3 II conjugation after irradiation in both U373 and LN229 cells.
Next, we investigated whether the inhibition of autophagy is affect reduced colony-forming ability after irradiation. We performed a colony-forming assay after transfecting cells with ATG5 siRNA (Fig. 7). The knockdown of ATG5 had no discernible effect on the clonogenic assay compared to the control (data not shown). However, ATG5 knockdown (RT+siRNA) significantly increased the colony number compared to RT (0.45 vs. 0.30 in U373, p<0.001; 0.57 vs. 0.40, p=0.005 in LN229) in both cell lines, suggesting that autophagy plays a role on cell death after irradiation (Fig. 7). The increased colony-forming ability with dexamethasone after irradiation was diminished after ATG5 knockdown in PTEN-mt cells. In U373 cells, the colony number of the RT+Dex was significantly reduced with siRNA treatment (0.543 vs. 0.49, p=0.01). However, in LN229 cel ls, the colony number of the RT+Dex+siRNA was not significantly different from that of the RT+Dex (0.63 vs. 0.65, p>0.05). Based on these results, we assumed that the dexamethasone-induced protection from cell death after irradiation may not be mediated by autophagy in PTEN functional cells.
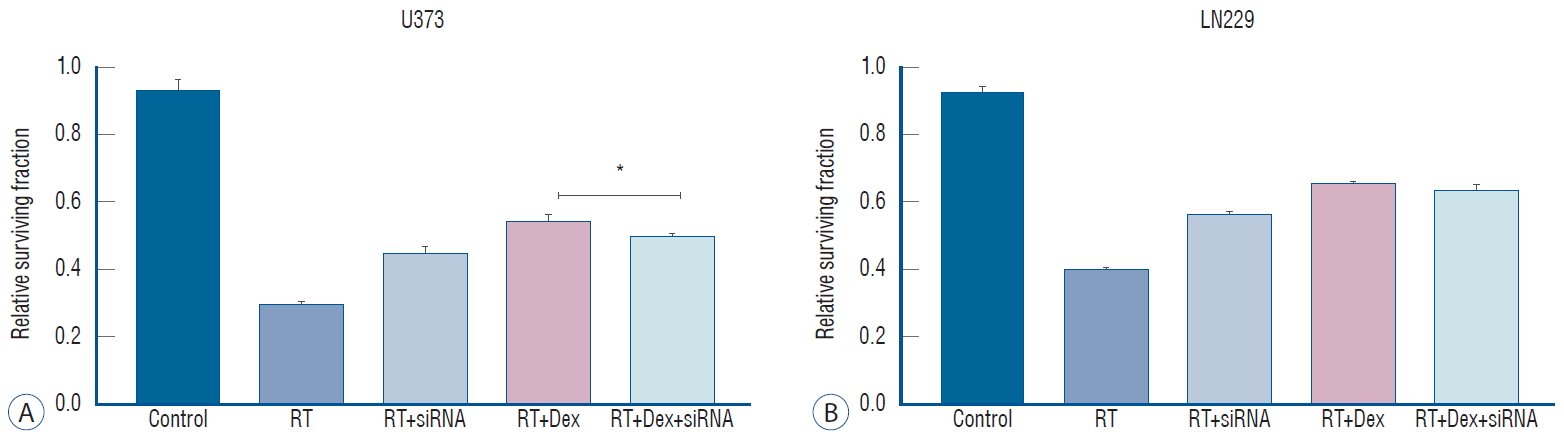
After autophagy-related gene 5 (ATG5) knockdown, the Dex effect of increased colony-forming ability after irradiation is significantly reversed in PTEN-mt U373 cells but not in PTEN-wt LN229 cells. *p<0.05. RT : radiation therapy, Dex : dexamethasone, siRNA : small interfering RNA.
DISCUSSION
In our study, we found out that dexamethasone increases cell viability against radiation effect in malignant glioma cells, which was related to radio-resistance. A selective phospho-AKT inhibitor (LY294002) resulted in the reversal of dexamethasone-induced increased cell survival after irradiation in PTEN-mt malignant glioma cells. Autophagy inhibition by ATG5 knockdown in these glioma cells resulted in the neutralization of dexamethasone-induced reduced cell death after irradiation not in PTEN-wt LN229 cells but in PTEN-mt U373 cells.
Dexamethasone reduces cell death of malignant glioma cells after irradiation
It is well-known that steroids act on glucocorticoid receptors and regulate the expression of downstream genes to exert its catabolic and anti-inf lammatory effects [26]. Beyond these hormonal effects, high-dose dexamethasone showed therapeutic effects against hematopoietic malignancy such as multiple myeloma and acute lymphoblastic leukemia [2,16]. In addition, steroids exert a lympholytic effect on the primary central nervous system lymphoma [38]. However, several studies have suggested that glucocorticoids may protect solid tumors from cytotoxic effects when used concurrently with radiotherapy or chemotherapy [9,21]. Using mouse model, Pitter et al. [28] suggested that glucocorticoid use during radiation therapy was controversial and may promote cell survival in glioblastoma. In a retrospective clinical study, the administration of dexamethasone during concomitant chemoradiotherapy with temozolomide resulted in poor prognosis in newly diagnosed GBM patients [30]. Recently, Luedi et al. [20] observed the effect of dexamethasone in patient-derived GBM stem cells. They found that when exposure to dexamethasone, isocitrate dehydrogenase 1 (IDH1) wild-type cells (typical of primary GBM) showed significantly higher invasion, cell proliferation, and angiogenesis than IDH1 mutant-type GBM stem cells. In addition, they evaluated the downregulation of apoptosis-related genes and the upregulation of oncogenic genes with dexamethasone through transcriptome microarray.
In our experiment, dexamethasone reduced cell death after irradiation in both malignant glioma cell lines of the common P53 mutation, which varied by PTEN functional status. However, the molecular mechanism of dexamethasone on glioma cells either directly or after binding to the glucocorticoid receptor remains largely unknown.
Dexamethasone affects autophagy differently in irradiated malignant glioma cells according to PTEN functional status
The uniqueness of our study was that we evaluated the effect of dexamethasone on irradiated malignant glioma cells in relation to autophagy, which has been known to be a response of tumor cells to relevant clinical doses of radiation [12]. In our previous study, we suggested autophagy as a molecular response that may lead to radiation-induced cell death or apoptosis [17].
Dexamethasone has been shown to induce autophagy in lymphocytes; however, the mechanism has yet to be fully elucidated [23]. In addition, autophagy activation was found to be secondary to dexamethasone-induced mitochondrial fragmentation in rat myoblast cells [35]. Wang et al. [37] suggested that dexamethasone inhibits autophagy in injured nerve cells in a dose-dependent manner.
PTEN acts as a classical tumor suppressor and is primarily involved in the homeostatic maintenance of the PI3K/AKT/mTOR cascade, which is a key regulator of autophagy [22]. PTEN is a known negative regulator of the PI3K/AKT/mTOR pathway, and PTEN expression has been found to increase autophagy in p53 mutant PTEN non-functional U87 mutant glioma cells [11]. When the effects of LY294002 on radiation were examined in the PTEN-mutant glioma cell line U251, low doses of the phospho-AKT inhibitor sensitized U251 cells to clinically relevant doses of radiation [25]. Andrade et al. [3] verified that dexamethasone suppresses PI3K-dependent AKT phosphorylation via glucocorticoid receptors in rat leukemic cells. Thus, we postulate that dexamethasone could be acting through glucocorticoid receptors to cause the effect observed in this study.
AKT is known to promote DNA damage repair, especially in irradiated cancer cells [34]. The PI3K/AKT/mTOR pathway negatively controls autophagosome formation, particularly in PTEN non-functional cancer cells. Although we observed the same increased G2/M arrest when irradiated glioma cells were pretreated with dexamethasone, the autophagy response to radiation was different according to PTEN status. In LN229 (PTEN functional glioma) cells, the increased G2/M arrest could induce a prolonged stressful condition to cell’s homeostasis that increased autophagy activity. However, in U373 (PTEN-non-functional glioma) cells, the dysregulated PI3K/AKT/mTOR pathway may quench the stress from increased G2/M arrest into reduced autophagy as AKT phosphorylation becomes upregulated in response to radiation damage.
In this study, we observed autophagy based on acridine orange staining, western blot, and fluorescence GFP-LC3 after treating cells with dexamethasone and irradiation. Our results were in accordance with different PTEN status of tested malignant glioma cells. In U373 PTEN-mutant cells, dexamethasone inhibited autophagy after radiation; however, in PTEN wild-type LN229 cells, it increased autophagy. In addition, the phospho-AKT inhibitor LY294002 increased autophagy in PTEN-mutant U373 cells but had no effect in PTEN wildtype LN229 cells. Thus, we can postulate that dexamethasone’ s effect on autophagy is mediated by phospho-AKT only in PTEN-deregulated pathways, not in PTEN functional cells.
In our study, the knockdown of ATG5 neutralized the effect of dexamethasone on autophagy after irradiation in both p53 mutant glioma cells having different PTEN status. However, the neutralization of dexamethasone’s effect for protecting from radiation-induced cell death was only observed in PTEN-deregulated U373 cells. ATG5 has been characterized as a protein specifically required for autophagy; it also has a role in the formation of autophagosomes. In addition, ATG5 can be a switch molecule of cross-talk between autophagy and apoptosis as the calpain-mediated cleavage of ATG5 has proapoptotic properties [42].
The molecular mechanism of dexamethasone-induced protection from cell death after irradiation in PTEN functional LN229 remains obscure in our study. We can only suggest the radiation-induced autophagy is not directly related to cell death in this circumstance. The catabolic effect of glucocorticoids can be responsible for the increased autophagy in PTEN functional glioma cells due to another nutrient sensing pathway such as AMPK or lysosomal activation. AMPK, a cellular energy activator/sensor, is a known negative regulator of mTOR pathway [14]. However, it is beyond the scope of this study. At this point and should be explored in a subsequent experiment.
Possible mechanism of action of dexamethasone in irradiated glioma cells
In our previous study, we observed that a lethal dose of radiation can induce autophagy and that G2/M fraction accumulates in proportion to the radiation dose [17]. Thus, we can suggest that DNA damage provoking G2/M arrest after irradiation was strong enough to increase autophagy in glioma cells.
Tusher et al. [36] identified a major change of gene expression after radiation using microarray in lymphoma cell lines. Among significantly upregulated genes after irradiation, 60% were genes related to cell cycle regulation, and 9% were involved in apoptosis. G2/M arrest activates corresponding cyclin-dependent kinases, which in turn activate repair machinery of DNA double-strand break, a common radiation injury [5]. In our study, increased G2/M arrest and decreased subG1 fraction with dexamethasone pretreatment may be a result of increased DNA double-strand break repair. Aasland et al. [1] had demonstrated that O6-methylguanine-DNA methyltransferase (MGMT), which is a significant biomarker for glioblastoma concomitant chemoradiotherapy with temozolomide, was induced by glucocorticoids because the MGMT promotor site has two glucocorticoid response elements (GRE) in HeLa S3 cells. Later, they evaluated that MGMT expression is not affected by radiation or temozolomide and is primarily controlled by specificity protein 1 (SP1) transcription factor in various malignant glioma cell lines. In addition, they suggested that p53 activation after irradiation suppresses its downstream target SP1, elevating the important role of glucocorticoid on MGMT expression via GRE binding in p53 mutant glioma cells.
Unfortunately, we neither quantified DNA damage after irradiation nor evaluated the expression of corresponding cyclin-dependent kinases. Thus, we could not say whether the dexamethasone protection from radiation-induced cell death in our study was from decreased DNA damage or increased DNA damage repair. We need to evaluate this point in future experiments, which will be dedicated to dexamethasone-induced DNA repair machinery change and responsible molecular pathway.
Conclusion
In our study, PTEN mutational status of malignant glioma cells is responsible for the different autophagy interference of dexamethasone after radiation. Autophagy inhibition after ATG5 knockdown reduces cell death or apoptosis only in PTEN non-functional glioma cells. Dexamethasone interference of autophagy observed in malignant glioma cells during our experiment seems to be partly mediated through the PI3K/AKT/mTOR pathway in PTEN-mt cells.
Our findings demonstrated that the combined treatment of dexamethasone with radiation in malignant glioma cells reduces cell death, which could be responsible for the radio-resistance frequently observed during radiotherapy in patients with malignant gliomas. Careful use of dexamethasone during radiotherapy could be important to minimize treatment failure in glioblastoma patients.
Notes
No potential conflict of interest relevant to this article was reported.
INFORMED CONSENT
This type of study does not require informed consent.
AUTHOR CONTRIBUTIONS
Conceptualization : JHI, HSG, KYL, JHK, BCY, HSC, JBP, JWK, SHS, HY
Data curation : AK, HSG, KYL
Formal analysis : AK, JHI, HSG, KYL, HSC
Funding acquisition : HSG, JHK, BCY, JBP, JWK
Methodology : AK, JHI, HSG, KYL, JHK, BCY, HSC, JBP, JWK
Project administration : HSG, JBP, JWK, SHS, HY
Visualization : AK, JHI, HSG, KYL, HSC, JBP
Writing - original draft : AK, JHI, HSG, KYL, HSC
Writing - review & editing : AK, JHI, HSG, KYL, JHK, BCY, HSC, JBP, JWK, SHS, HY
Acknowledgements
A part of this study was presented at the 13th Winter Meeting of Korean Brain Tumor Society (2019 January 25).
This work was supported by grants from National Cancer Center, Korea (NCC1710871-3 and 1910090-2), and a grant of the Korea Health Industry Development Institute, funded by the Ministry of Health & welfare, Republic of Korea (HI17C1018).
The first author (A.K.) sincerely thanks the Korea Foundation for International Healthcare (KOFIH) and the Uganda Cancer Institute (UCI) for giving him the opportunity to perform this study and supporting him throughout his training program.
Supplementary Materials
The online-only data supplement is available with this article at https://doi.org/10.3340/jkns.2019.0187.
Supplementary Fig. 1
Cell survival curve with or without dexamethasone. The addition of dexamethasone at the concentration used in this experiment (10 μM) has no effect on cell proliferation compared to the control.
Supplementary Fig. 2.
Akt phosphorylation in response to radiation is observed in U373 cells but not in LN229 cells. Akt : protein kinase b.
Supplementary Fig. 3.
A : The efficiency of ATG5 knockdown by siRNA was confirmed by western blot. b : ATG5 knockdown decreases autophagy after irradiation in both U373 and LN229 cells. ATG5 : autophagy-related gene 5, siRNA : small interfering RNA.