Pediatric Glioma at the Optic Pathway and Thalamus
Article information
Abstract
Gliomas are the most common pediatric tumors of the central nervous system. In this review, we discuss the clinical features, treatment paradigms, and evolving concepts related to two types of pediatric gliomas affecting two main locations: the optic pathway and thalamus. In particular, we discuss recently revised pathologic classification, which adopting molecular parameter. We believe that our review contribute to the readers’ better understanding of pediatric glioma because pediatric glioma differs in many ways from adult glioma according to the newest advances in molecular characterization of this tumor. A better understanding of current and evolving issues in pediatric glioma is needed to ensure effective management decision.
INTRODUCTION
Gliomas are the most common central nervous system (CNS) tumors affecting pediatric patients [60]. These tumors exhibit an extremely broad range of clinical behaviors. Their clinical features and courses can be affected by various factors, particularly location and pathology. The site of origin of a pediatric glioma within the CNS [60] can influence both the clinical presentation and the selection of surgical management. Various pathological subgroups of pediatric glioma, ranging from indolent (e.g., pilocytic astrocytoma) to aggressive tumors (e.g., glioblastoma), have been identified. The pathological characteristics of these subgroups should be considered during tumor management.
In the last decade, significant progress has been made in our understanding of the molecular biology underlying pediatric gliomas [11,42,81]. Notably, biologic subgroups characterized by distinct molecular, pathologic, and clinical features have been identified, with the 2016 World Health Organization classification of tumors of the CNS adopting this concept [55]. In this review, we summarize the literature regarding pediatric gliomas located in the supratentorial deep midline in terms of both current concepts and evolving issues. We particularly focus on two main locations, the optic pathway and thalamus. Accordingly, our review is constructed around the two main themes of gliomas occurring in these locations.
OPTIC PATHWAY GLIOMAS
Clinical features
Optic pathway gliomas (OPGs) account for approximately 3–5% of pediatric brain tumors [8,72]. Approximately 75% of these tumors are diagnosed during the first decade of life, with 60% of patients receiving a diagnosis before the age of 5 years; the incidence does not differ by sex [8,26]. OPGs can affect several anatomic regions along the optic pathway. The size and extent of the tumor influences the presentation, ranging from pure optic pathway gliomas (which are located in the optic nerve, optic chiasm, optic tracts and, less frequently, optic radiation) to larger exophytic tumors with extension into the hypothalamus, pituitary gland, third ventricle, and diencephalon [8,26]. Overall, 25% of OPGs are confined to the optic disc and nerve, whereas 40–75% involve the chiasm; of the latter, 33–60% are considered posterior lesions and also involve the hypothalamus or third ventricle [8].
Patients with OPGs may be asymptomatic or may present with variable symptoms, depending on the tumor location. The presenting symptoms are mainly ophthalmologic, including decreased or loss of vision, proptosis, strabismus, and nystagmus; these symptoms are often caused by a compromise of the optic pathways or diencephalon by either direct tumor invasion or compression [4,26,36]. Non-ophthalmologic symptoms may include endocrine dysfunction, cognitive problems, and behavioral disturbances [4,26]. Endocrinologic symptoms such as precocious puberty and diencephalic syndrome indicate that an OPG affects the midline hypothalamic structures [72]. Symptoms of increased intracranial pressure (ICP) with hydrocephalus may also occur if the tumor obstructs the flow of cerebrospinal fluid through the third ventricle [26].
Incidentally discovered OPGs are typically associated with neurofibromatosis type 1 (NF-1). The reported incidence of NF-1 among patients with OPGs varies from 10 to 70%; a majority of patients with these lesions are thought to have NF-1 [26,67,72]. OPGs associated with NF-1 are more often benign, may be multifocal and bilateral, and are usually found within the optic nerve [31,45,82]. Notably, the presence of bilateral OPGs is a pathognomonic feature of NF-1 and was identified in 34.8% of patients with the condition. Chiasmatic glioma is rarely associated with NF-1; however, such tumors often have a more aggressive course with diencephalic syndrome, and typically progress [31,45,82].
Before making treatment decisions, as much information as possible must be obtained from imaging evaluations. The diagnoses of OPGs with typical imaging appearances can be confirmed using modern imaging modalities and protocols [8,26]. Radiologic evaluations of the tumor and surrounding structures play a critical role in pre-surgical planning. Although both computed tomography and magnetic resonance imaging (MRI) are useful for radiologic evaluations, the latter is considered the mainstay of diagnostic imaging for pediatric OPGs [26]. In such cases, typical MRI findings include the appearance of an iso- to hypointense lesion on T1-weighted images, hyperintensity on T2-weighted images, and homogeneous enhancement with Gd administration [8,78]. These tumors are normally solid but may have cystic components; however, they rarely exhibit leptomeningeal spread, which tends to occur with more aggressive tumors such as pilomyxoid astrocytomas [26,44,55]. Therefore, imaging can be used for tumor classification. Several researchers invented radiological classification using OPGs’ anatomic location and their relation to the optic pathways [21]. A recent report further proposed an MRI interpretation protocol intended to reduce intraobserver discrepancies [47]. We note, however, that prognostic validation is needed for classifications based solely on radiologic features, as these do not address surgical, genetic, and pathological factors.
Recently, newer MR imaging techniques such as MR spectroscopy (MRS), perfusion and diffusion-weighted MRI, and diffusion tensor imaging have been used for the pre- and posttreatment evaluation of OPGs. Reports have described the use of the former two techniques for tumor grading [71,87], whereas the latter has been used to assess the integrity of white matter tracts, including the optic radiations [72,76].
Management
Pathologically, OPGs are generally considered low grade [8,26]. Juvenile pilocytic astrocytoma is the predominant histology and high-grade malignant pediatric OPGs are infrequently observed. Although clinical course predictions are difficult, as the natural courses range from stable disease to progression and even spontaneous regression [34,59,65,76]. However, a large proportion of OPGs exhibit an indolent nature, particularly in the context of NF-1 [59]. Still, tumor progression is typically observable within 2 years of diagnosis [83]. Despite the rarity of progression after the age of 12 years, some patients may lose their vision in adolescence. Therefore, these children should be subjected to careful vision monitoring until they reach adulthood.
Currently, the management options for OPGs include followed observation, chemotherapy, radiation therapy, and surgery. Given the pathologic and genetic diversity of OPGs, good long-term survival and functional results require a multidisciplinary approach. Careful observation and timed intervention may be appropriate and reasonable options for these generally indolent tumors, particularly in the context of NF-1. However, patients presenting with symptoms and/or tumor progression on serial imaging scans should be considered candidates for appropriately selected interventions.
Because a significant portion of patients with OPGs also have NF-1, we consider it important to discuss the current recommendations for the screening of this population of patients. In 1997, the NIH NF-1 Optic Glioma Task Force outlined recommendations for the screening, follow-up, and treatment of OPG in children with NF-1 [53]; Listernick et al. [52] updated these initial recommendations in 2007. The updated recommendations strongly emphasize an annual screening examination, including visual acuity testing, confrontation visual field testing, color vision, pupillary reflexes, and an assessment of eyelids, ocular motility, and fundi, for children up to 8 years of age. Although no agreement has been reached regarding the time interval for the screening of patients older than 8 years, the guidelines recommend biennial screening until the age of 18 years until proven otherwise. Furthermore, the guidelines do not recommend using visual evoked potential as a screening tool. Regular ophthalmological examinations of NF-1 children with a known OPG at 3-month intervals are recommended during the first year, with larger intervals thereafter if progression is not evident.
OPGs require an individualized treatment approach based on combinations of surgery, chemotherapy, and/or radiotherapy. Accordingly, management decisions should be made through multidisciplinary team discussion after weighing the pros and cons of each treatment. For many years, the treatment of choice for OPG was surgical excision, which provided either a cure or prolonged stability [32,84]. However, in some cases, such as NF-1 patients, it is discouraged because it inevitably causes blindness on the affected side, with the risk of bilateral loss of vision [49]. Furthermore, surgical biopsy has been supplanted by imagingassisted virtual diagnosis. Therefore, surgery should only be used in specific circumstances, such as the treatment of lesions causing significant unilateral visual compromise (often blindness) with evidence of extension into the chiasm, which thus threatens vision in the other eye. Here, prechiasmatic transection of the optic nerve can prevent tumor progression toward the chiasm, thereby maintaining vision in the contralateral eye [9]. In such cases, thorough clinical and radiologic testing should be used to ensure that there is no involvement of the chiasm prior to surgical intervention. Other circumstances, including painful proptosis, corneal exposure, and disfigurement with severe visual loss in the involved eye, may also warrant surgical intervention [49,52,72]. Additionally, significant OPG growth can compress the third ventricle and thus cause hydrocephalus, necessitating lesion debulking to prevent further neurologic compromise [4,26].
Chemotherapy has become the predominant treatment modality for OPGs. Previously, this option was recommended as a first-line treatment for children under 7 years of age. Older children were initially offered radiotherapy [8], as chemotherapy was shown to delay the need for radiotherapy in young children and thus allow time for neuropsychological development. However, the avoidance of radiation toxicity has led to the emergence of chemotherapy as the preferred first-line treatment, regardless of age, since 2000 [36,54]. Chemotherapy may stabilize or reduce the disease burden and thus prevent the need for neurosurgery and radiotherapy [26,36,57]. Although no single ideal chemotherapeutic agent has been identified, the current standard chemotherapy regimen of vincristine and carboplatin, commonly known as the Packer regimen [62,63], was reported by Packer and colleagues to yield progression-free survival rates of 75% at 2 years and 68% at 3 years [62]. Alternative chemotherapeutic regimens including thioguanine, procarbazine, lomustine, and vincristine with or without dibromodulcitol have yielded efficacies similar to that of the classical carboplatin and vincristine regimen [3,68]. Temozolomide has also been shown to stabilize disease in more than 50% of patients without significant toxicity, and is considered an option for patients with progressive OPG in whom first-line therapy has failed [33]. Additionally, a recent report described improved visual function in select patients treated with bevacizumab [5]. A systematic review of the effects of chemotherapy on visual outcomes found that less than 15% of cases demonstrated improvement, while 40% exhibited deterioration. Overall, the investigators concluded that chemotherapy did not improve the visual outcomes of children with OPGs [58]. However, among NF-1 patients treated with chemotherapy, visual acuity remained stable or improved in 72% of patients and declined in 28% [24]. Researchers have also highlighted the importance of earlier treatment (i.e., treatment initiation <5 months from diagnosis) to prevent the occurrence of irreversible vision damage [2,24].
The role of radiotherapy in OPGs has changed considerably over the past decade. It used to be the treatment of choice, ahead of chemotherapy [15]. Radiotherapy is an effective treatment for OPGs, with reported 10-year rates of progression-free survival as high as 90% [80]. Conventional external beam radiotherapy for OPG is most commonly administered at total doses of 45–60 Gy in 1.6–2.0 Gy fractions [27,80]. However, its use has been limited by significant risks, including neuroendocrine dysfunction, and neurodevelopmental delays [27,33]. Patients with NF-1 are particularly at risk of secondary neoplasms following radiation [77]. Radiation damage also increases the risks of a secondary exacerbation of visual loss, Moyamoya syndrome, and cerebro-occlusive disease [28]. Approximately 3.5% of all children receiving radiation therapy will develop Moyamoya syndrome; patients with NF-1 have a much higher risk of this condition, compared to the general population [20]. Therefore, radiotherapy is now reserved for tumors refractory to chemotherapy and surgery.
Fractionated stereotactic radiotherapy, proton beam radiotherapy, and stereotactic radiosurgery have been evaluated as means of reducing the adverse effects of conventional radiotherapy. The former is highly efficacious, with a progression-free survival rate of 72% at 5 years [18]. Proton and photon beam therapy, which administer lower doses of radiation to the temporal lobes and pituitary gland, have been tested in small series of patients with promising results [51]. Finally, Gamma knife radiosurgery (single session) has been used to treat optic pathway and hypothalamic gliomas; in a recent case series, this modality yielded a progression-free survival rate of 83% at 3 years, and more than 80% of patients experienced stabilized or improved visual acuity [22]. Despite the efficacies of these new radiotherapy modalities, physicians should remain cautious about the potential for serious adverse effects.
THALAMIC GLIOMAS
Clinical features
Although thalamic gliomas are rare, accounting for 1–5% of pediatric intracranial tumors [37,46,79], the incidence of thalamic glioma is relatively high in children relative to adults (1%) [37]. Pediatric patients are affected at a median age of 8–10 years, without sex predilection [29,37,79]. The duration of symptoms varies in children, although the mean value of 6 months [37] is longer than that in adults. However, a shorter symptom duration may portend a worse prognosis and may correspond to a higher pathologic grade.
Children with thalamic gliomas may present with a range of symptoms attributable to the deep locations of these tumors [37,70]. The most common symptoms include an increased ICP, headache, lethargy, nausea, vomiting and, if left untreated, stupor and coma. Visual symptoms are present in as many as 50% of cases. Motor deficits are common, whereas sensory disturbances and movement disorders are less common. In addition to the deep location, tumor effects on the structures surrounding the thalamus may lead to several symptom complexes. For example, hypothalamic involvement results in endocrinopathies, whereas mammillothalamic tract or fornix involvement can cause cognitive dysfunction and memory problems. Additionally, bilateral thalamic lesions manifest the characteristic but unusual presentation of personality changes, memory loss, confusion, hallucinations, hyperphagia, and bradyphrenia [37].
Thalamic gliomas can be categorized by growth characteristics as focal, diffuse, or bilateral [37]. Focal tumors occur at very discrete locations within the thalamus and grow to expand beyond the anatomical boundaries into the surrounding white matter and/or ventricles (third and lateral). These tumors typically correspond to World Health Organization (WHO) grade I juvenile pilocytic astrocytoma [12], and can be further subclassified according to the brain-tumor interface or a cystic or noncystic component. In contrast, the diffuse type does not display a well-demarcated tumor interface and thus typically migrates beyond its anatomical boundaries. These tumors typically correspond to the WHO grade II–IV fibrillary type [13].
Bilateral-type thalamic gliomas merit a separate discussion. These tumors, which have an approximate incidence of 14%, are presumed to have a de novo bilateral origin, like multifocal gliomas, and exhibit bilateral symmetry at the time of diagnosis [79]. As in other reports, patients with bilateral thalamic glioma had a relatively shorter mean duration of symptoms (2.5 vs. 8.7 months for unilateral thalamic gliomas) [79]. Surprisingly, although biopsies of these lesions have indicated a lower histologic grade, the prognosis is much worse; in other words, these tumors have a malignant course despite the lower-grade histology [37,79].
Radiologic features provide significant clues about the histology of and management options for thalamic gliomas. Advanced neuroimaging provides additional information about the metabolism and physiology of these lesions and thus contributes to diagnosis and follow-up [10,46]. Similar to the clinical presentations, the imaging characteristics of these tumors vary depending on the grade of malignancy. Up to 20% of tumors are solid and cystic with calcification [10]. Furthermore, although most malignant gliomas exhibit peritumoral edema, enhancement, and necrosis, these characteristics may be absent from some high-grade tumors, which may reduce the reliability of tumor classification and grading based on conventional imaging [64]. Advanced MRI techniques, such as perfusion MRI and MRS, can provide additional information to support tumor classification. On perfusion MRI, low-grade tumors have a relatively low regional cerebral blood volume, compared to high-grade tumors [56]. On MRS, high-grade tumors such as glioblastoma exhibit a high choline peak with a low N-acetylaspartate peak and high choline-to-creatine ratios. Additionally, elevated levels of lipid and lactate, which correlate with necrosis, help to differentiate glioblastomas from high-grade gliomas such as anaplastic astrocytoma [10].
Management
The treatment regimen for pediatric thalamic glioma has not been clearly defined. Clinicians previously favored a conservative approach comprising stereotactic biopsy and radiotherapy over radical resection, given the risks of postoperative morbidity and mortality [16,25]. However, recent improvements in imaging modalities, neuronavigation systems, microsurgical techniques, and intraoperative electrophysiological monitoring have yielded clear anatomical pictures of these deeply seated lesions; therefore, radical resection of a selected subset of these tumors is associated with minimal morbidity and mortality [14,37,50,74]. Generally, patients who have undergone total or subtotal resections of thalamic tumors experience improved outcomes and survival prognoses compared to those who do not undergo surgery [85]. Improved overall survival is associated with the extent of resection and can be achieved with minimal occurrences of morbidity and mortality [85].
Contrast-enhancing discrete lesions with well-defined boundaries and no involvement of the posterior limb of the internal capsule are best suited for radical resection [74]. Various surgical approaches have been described for thalamic gliomas [37,74,79], although the position of the internal capsule relative to the tumor is a key factor when determining the approach. The optimal approach typically depends on the location of the tumor within the thalamus and the extent and relationship of the tumor to critical structures [79]. Bilginer et al. [7] favor the anterior interhemispheric transcallosal approach for superior thalamic tumors and the posterior interhemispheric parasplenial approach for posterior thalamic tumors. Ozek and Türe [61] also recommend the interhemispheric transcallosal approach for patients with superior and anterior thalamic tumors. In contrast, Baroncini et al. [6] prefer the transcortical frontal approach for tumors in the superior thalamus. For large tumors in highly functional areas or areas with poorly defined boundaries, Puget et al. [70] recommend the staged excision of thalamic tumors. Tumors that cannot be resected safely should be investigated using biopsy sampling to exclude non-neoplastic pathological entities and direct future adjuvant therapies based on histological tumor types. Stereotactic or endoscopic biopsy should be considered in cases that may require only a tissue diagnosis or are otherwise appropriate (e.g., bilateral thalamic tumors) [37]. For unilateral lesions with diagnostically ambiguous imaging findings, an imaging or endoscopically guided biopsy can establish a diagnosis and allow the identification of molecular markers before initiating an open approach [37].
Radiotherapy and chemotherapy should be reserved for patients with malignant thalamic lesions, multiple recurrent tumors, or residual disease [37]. However, neither option is superior to complete resection, and both carry potential long-term adverse effects that are of significant concern in young children. Therefore, adjuvant therapies should not be considered without a histological diagnosis [37]. For incompletely resected or progressive lesions, most centers administer a total radiation dose of 54 Gy administered over 30 fractions [35,37,66]. However, many centers are switching to primary chemotherapy to avoid late irradiation effects such as endocrinopathies, vasculopathies, optic nerve damage, and loss of intellectual function. Therefore, reoperation should be offered, if feasible, to address a small residual, recurrent, or regrown low-grade glioma on serial imaging.
Currently, no standardized protocol exists for the chemotherapeutic treatment of low-grade gliomas. Generally, these lesions are treated with a combination of agents, although studies have reported varying levels of success with single agents [35]. However, a combination of vincristine and carboplatin and vinblastine monotherapy have been accepted as first-line adjuvant chemotherapy regimens [35]. In contrast, the standard treatment for high-grade gliomas comprises surgery followed by radiotherapy. Although various combinations of chemotherapeutic agents have been tested, adult studies have led most pediatric neuro-oncology groups to adopt the combination of radiotherapy with concomitant and adjuvant temozolomide as the standard of care [23,35].
The factors influencing survival among patients with thalamic glioma include the tumor grade, extent of resection, adjuvant therapy, and symptom duration [7,46,79]. Although children with low-grade gliomas have relatively good overall survival rates, the thalamic location portends a worse outcome, despite otherwise similar tumor biology [37,85]. For high-grade gliomas, radical surgical resection remains the most important prognostic factor and should be considered unless severe morbidity is anticipated [14,74,85]. Given their location, bilateral thalamic tumors are considered inoperable beyond diagnostic biopsy [79], and have been reported to be less responsive to radiotherapy and chemotherapy treatment relative to unithalamic tumors [37,79]. Consequently, bilateral thalamic tumors are associated with a poor outcome.
CONTROVERSIAL AND EVOLVING ISSUES
Several currently evolving issues in this field are notable. First, the 2016 World Health Organization Classification of Tumors of the CNS (Table 1) includes molecular parameters, as well as histology, in the definitions of CNS tumor entities [55]. This represents a significant break from the paradigm of the past century, wherein brain tumor classification was based largely on the concept of histogenesis; that is, tumors can be classified according to their microscopic similarities to different putative cells of origin and their presumed levels of differentiation [55]. Studies conducted over the past two decades have clarified the genetic basis of tumorigenesis in brain tumor entities, raising the possibility that such an understanding may contribute to the classification of these tumors [11,42,81]. Accordingly, the inclusion of molecular parameters in the 2016 WHO classification has led to significant changes in the diagnosis of glioma. Diffuse glioma now includes WHO grade II and grade III astrocytic tumors, grade II and III oligodendrogliomas, grade IV glioblastomas, and the related pediatric diffuse gliomas. Accordingly, this approach distinguishes diffuse gliomas from astrocytomas that have a more circumscribed growth pattern, lack IDH gene family alterations, and frequently harbor BRAF alterations (pilocytic astrocytoma, pleomorphic xanthastrocytoma) or TSC1/TSC2 mutations (subependymal giant cell astrocytoma). Accordingly, as diffuse astrocytoma and oligodendrogliomas are now considered nosologically more similar than diffuse astrocytoma and pilocytic astrocytoma, the family trees have been redrawn.
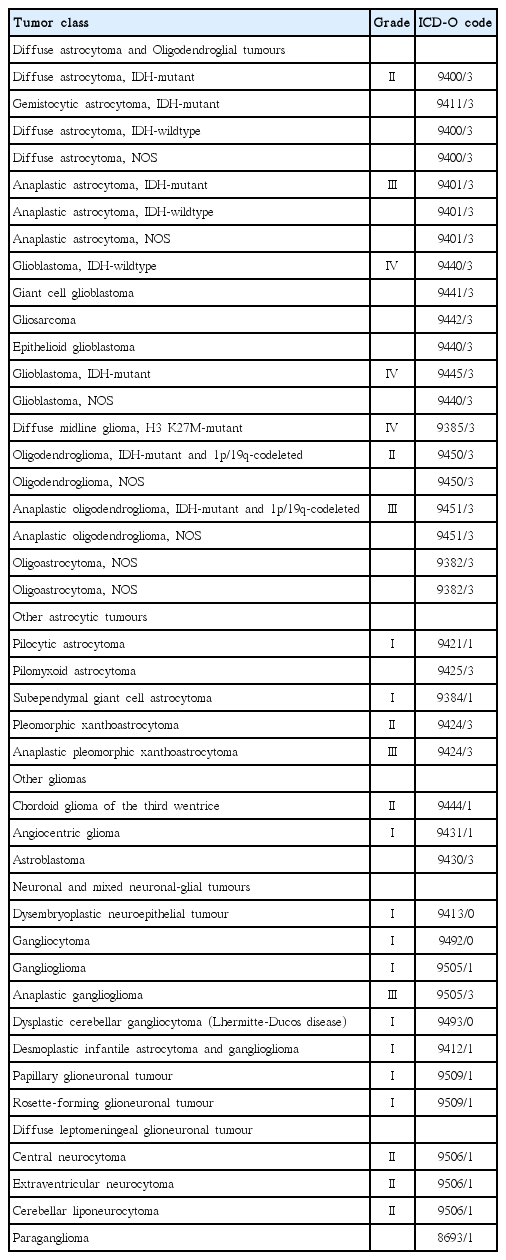
Moreover, despite histological similarities, the molecular characteristics of pediatric gliomas differ greatly from their adult counterparts, both in terms of mutation patterns and the prognostic implications of those mutations. These discoveries include the identification of recurrent mutations, fusion, and duplication events in the BRAF, FGFR1, MYB, and MYBL1 genes in pediatric low-grade gliomas, as well as mutations affecting histone components (H3F3A p.K27M or p. G34R/V) in pediatric high-grade gliomas [11,42,73]. Adult low-grade gliomas are characterized by IDH1/2 mutations and ATRX mutations in astrocytic tumors and IDH1/2 mutations and 1p/19q codeletions in oligodendroglial tumors. TERT promoter mutations are also noted in low-grade gliomas and are mainly associated with oligodendrogliomas [73].
Second, the concept of pediatric diffuse glioma has evolved. Previously, pediatric diffuse gliomas were grouped with their histologically similar adult counterparts, despite known differences in behavior. The group of tumors that primarily occur in children can be genetically characterized by K27M mutations in the histone H3 gene H3F3A (approximately 75% of cases) or, less commonly, in the related HIST1H3B gene (approximately 25%) and exhibit clinically similar diffuse growth patterns and midline locations (e.g., thalamus, brain stem, and spinal cord). This newly defined entity is termed diffuse midline glioma, H3 K27M–mutant, and includes tumors previously referred to as diffuse intrinsic pontine glioma (DIPG) [55].
Third, the grading of pilomyxoid astrocytoma has also evolved. In our experience, the pilomyxoid variant always follows a more aggressive course when compared to the suprasellar pilocytic astrocytoma, which has a more classic appearance. Accordingly, a suggestion was made to suppress the grading of pilomyxoid astrocytomas, which would be automatically assigned to WHO grade II until further studies have clarified their behavior.
These advances in pathological diagnosis have led to significant changes in routine clinical practice. For example, tumor genetic profiling has become an essential part of an evaluation. Along with this, the phenotypical and molecular identification of tumors can guide therapies directed against the effects of these mutations. At present, the mainstay of pediatric glioma therapy is surgical treatment, which may be curative for some low-grade gliomas when total resection is possible. However, when total resection is not possible or in cases of high-grade gliomas, the chances for progression or relapse are substantial. Although current adjuvant chemotherapy and radiotherapy are associated with relatively good overall tumor control in lowgrade gliomas, they have shown unsatisfactory results for highgrade gliomas. Even for low-grade gliomas, long-term treatment with adjuvant therapy is often associated with significant morbidity [1]. Thus, a more tailored and novel approach is needed for pediatric gliomas.
Another interesting discovery is the anatomic distribution of histone mutations in pediatric high-grade gliomas. K27M mutations are predominantly found in tumors arising in midline locations such as the thalamus, pons, and medulla oblongata. Histone H3.3 G34R or G34V mutations and BRAFV600E mutations are predominantly found in hemispheric tumors. Other gene alterations associated with histone gene mutations are also found in location-specific patterns. For example, ATRX/DAXX mutations are associated with cortical G34R/V tumors [40,81]. Furthermore, diffuse midline gliomas, H3 K27M-mutant (WHO grade IV) are a distinct group that was introduced in the recent revision of the WHO classification [55]. This group includes the tumors that were previously referred to as DIPG, as well as approximately 50–60% of diffuse gliomas that arise in the thalamus or spinal cord [75,86]. Current treatment of these tumors does not include surgical resection due to their challenging location. However, several academic centers around the world have recently incorporated upfront biopsy for the identification of potential molecular alterations for targeted therapy [69]. The surgically inaccessible location of these tumors, along with the fact that diffuse midline glioma H3 K27M-mutant is less chemosensitive and radiosensitive, results in a poor prognosis for affected children, who have a median survival time of 9–12 months and a survival rate of less than 10% two years after initial diagnosis, regardless of histological grade [17,43]. In comparison, children with histone wild-type diffuse midline gliomas show significantly better overall survival times (mean, 4.5 years) [43].
Additionally, in patients with NF-1 associated OPGs, there is inactivation of the NF-1 tumor suppressor gene. The NF-1 gene located on chromosome 17q, which encodes neurofibromin, a negative regulator of RAS proto-oncogene [30]. The NF-1 gene regulates cell differentiation, proliferation, and survival through mitogen activated protein kinase and mammalian target of rapamycin (mTOR) pathways [41]. Dysfunction of the NF-1 gene has been found to affect the mTOR pathway promoting cell survival, growth, and proliferation [19]. And, it also activates RAS resulting in unregulated cell proliferation [48]. In the case of sporadic OPGs, it demonstrates a rearrangement in the kinase portion of the serine/threonin-protein kinase BRAF gene caused by fusion with KIAA1549 because of a duplication at 7q34 resulting in activation of BRAF [38,39].
Several efforts are underway to develop novel therapeutic approaches for the treatment of pediatric gliomas, but significant challenges remain in translating these insights into clinical practice. Nonetheless, new molecular therapeutics are being evaluated for the treatment of pediatric gliomas, including crizotinib (a MET-fusion target), dabrafenib (a BRAF inhibitor), trametinib (a MEK inhibitor), nimotuzumab (an immunoglobulin G1 antibody that targets EGFR), crenolanib and dasatinib (a PDGFR inhibitor), and bevacizumab (an angiogenesis inhibitor) [11,40]. Many clinical trials investigating these targeted agents are still in the early phases, so the outcomes are pending. The success of targeted and personalized therapy not only requires these results, but also continued research into basic signaling mechanisms and the gene regulatory networks that drive the biology of pediatric brain tumors.
Notes
No potential conflict of interest relevant to this article was reported.
INFORMED CONSENT
This type of study does not require informed consent.
Acknowledgements
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2017R1C1B5018208).