Medulloblastoma in the Molecular Era
Article information
Abstract
Medulloblastoma is the most common malignant brain tumor of childhood and remains a major cause of cancer related mortality in children. Significant scientific advancements have transformed the understanding of medulloblastoma, leading to the recognition of four distinct clinical and molecular subgroups, namely wingless (WNT), sonic hedgehog, group 3, and group 4. Subgroup classification combined with the recognition of subgroup specific molecular alterations has also led to major changes in risk stratification of medulloblastoma patients and these changes have begun to alter clinical trial design, in which the newly recognized subgroups are being incorporated as individualized treatment arms. Despite these recent advancements, identification of effective targeted therapies remains a challenge for several reasons. First, significant molecular heterogeneity exists within the four subgroups, meaning this classification system alone may not be sufficient to predict response to a particular therapy. Second, the majority of novel agents are currently tested at the time of recurrence, after which significant selective pressures have been exerted by radiation and chemotherapy. Recent studies demonstrate selection of tumor sub-clones that exhibit genetic divergence from the primary tumor, exist within metastatic and recurrent tumor populations. Therefore, tumor resampling at the time of recurrence may become necessary to accurately select patients for personalized therapy.
INTRODUCTION
Medulloblastomas are heterogeneous, highly aggressive tumors of the central nervous system and are the most frequent malignant brain tumors in children [14,16,67]. Most medulloblastomas are sporadic and arise in the posterior fossa due to deregulation of cerebellar development [34]. In rare cases, medulloblastoma can be associated with inherited disorders such as Li-Fraumeni, Turcot or Gorlin syndrome [23,52].
Integrative genomic studies from several independent research groups have shown that medulloblastoma is not a single disease but is comprised of at least four subgroups with specific demographic, genetic, transcriptional, clinical, and prognostic characteristics [31,43,62,66,76,85,89]. The medulloblastoma subgroups are termed wingless (WNT), sonic hedgehog (SHH), group 3, and group 4 and were agreed upon by experts from around the world during a consensus meeting in Boston in 2010 [85].
The 5-year overall survival of medulloblastoma patients is 60–70% under the current standard multimodal treatment consisting of maximal safe tumor resection, chemotherapy and, for non-infant (>3–5 years) patients, craniospinal irradiation [20,87]. Unfortunately, improved outcome has been associated with serious long-term treatment sequelae such as neurocognitive impairment, endocrine deficiencies, and secondary tumors [32,39,40]. During the last two decades, tumor staging of medulloblastoma patients has been solely based on clinical factors (patient’s age, presence or absence of metastases at diagnosis, postoperative residual tumor) and, in some studies, histopathological subtypes [85]. A recently proposed, refined risk stratification of non-infant medulloblastoma patients based on subgroup and outcome data allows a classification of patients into four groups with different prognoses : “low risk” (>90% survival), “standard risk” (75–90% survival), “high risk” (50–75% survival), and “very high risk” (<50% survival). This new approach to patient stratification opens the door for future clinical trials including treatment de-escalation for patients with favorable outcomes and development of urgently needed new therapies for patients with high-risk disease [61].
Leptomeningeal dissemination occurs in up to 40% of patients at time of diagnosis and almost all patients present with metastases at time of recurrence [96]. Despite good progress in the clinical management of patients with medulloblastoma, recurrent and metastatic disease remain incurable and metastatic relapse is the primary cause of death in children with medulloblastoma. Thus, characterization of the molecular mechanisms of metastatic spread and survival in the metastatic niche, coupled with the identification of targetable vulnerabilities in these processes is a key area of current and future investigation.
MOLECULAR SUBGROUPS
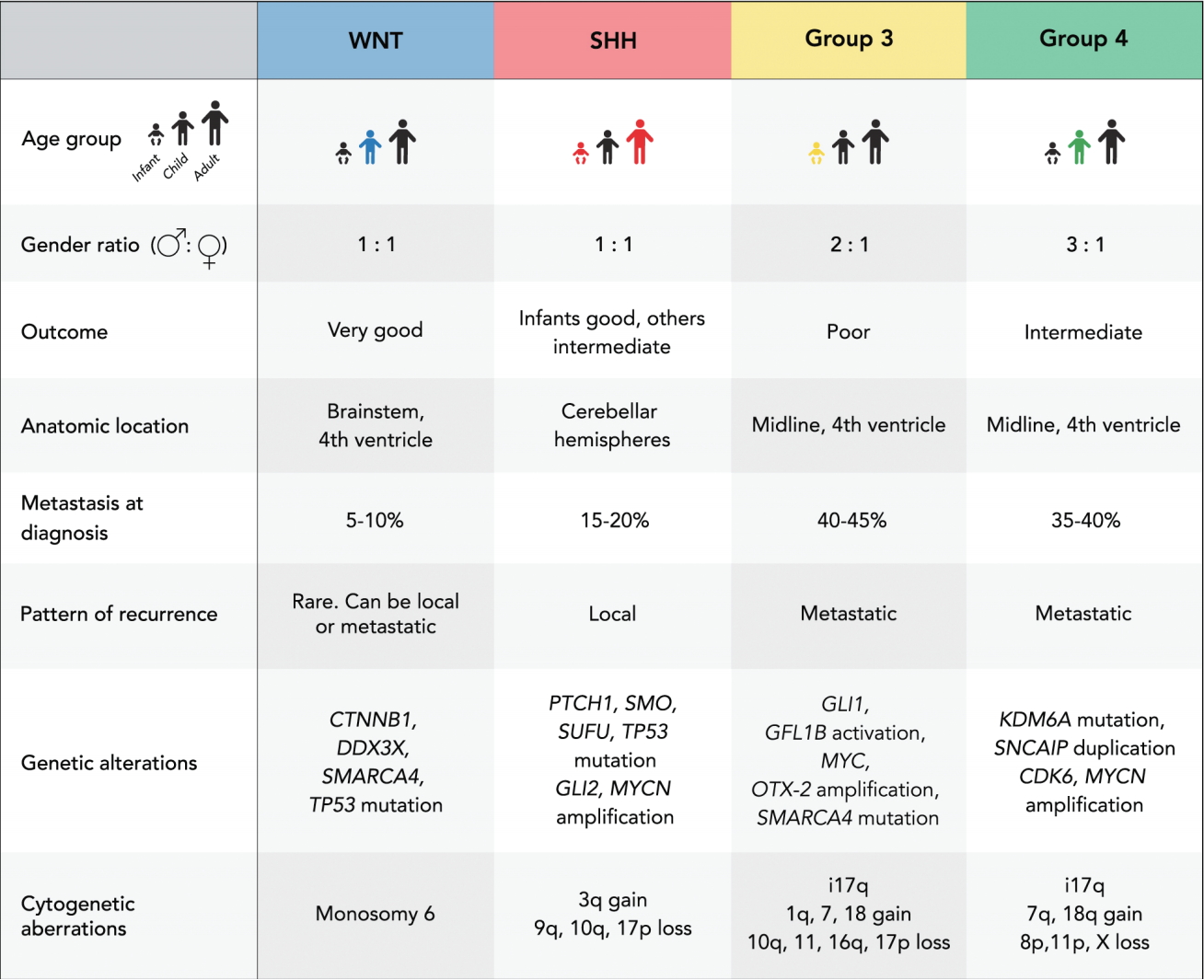
The four subgroups (WNT, SHH, group 3, and group 4) were identified based on integrated genomics studies and feature well-defined clinical, histopathological, genetic, transcriptional, and prognostic characteristics (Fig. 1) [45,46,57,62,63,70,73,76,85,95]. Recent research suggests that, based on genetic, transcriptional and epigenetic data, medulloblastoma can be divided even further into molecularly-determined subtypes which may potentially improve patient stratification in future clinical trials [6,42,50,74,75].
WNT
This represents the rarest subgroup and accounts for approximately 10% of all medulloblastomas. Children and adolescents are the most commonly affected age groups [27]. WNT medulloblastomas are thought to arise from progenitor cells of the dorsal brain stem in the lower rhombic lip and typically present with somatic mutations in the CTNNB1 gene which encodes beta-catenin and leads to an overexpression of the subgroup-defining WNT signaling pathway [16,44]. Monosomy of chromosome 6 is characteristic of this subgroup [42,46]. TP53, DDX3X, and SMARCA4 mutations have also been described in patients with WNT tumors [24,42,55,67,101]. WNT medulloblastomas are rarely metastatic and have a favorable outcome compared to the other subgroups [76].
SHH
A bimodal age distribution is typical for SHH tumors, with a peak incidence during infancy and adolescence [27]. About 30% of all medulloblastomas are classified as SHH tumors which are frequently located laterally in the cerebellar hemispheres [27,54]. There is evidence, that SHH medulloblastoma originates from cerebellar granule precursor cells of the external granule layer [8,16,44]. Hyperactivation of the SHH signaling pathway is characteristic of this subgroup and is often due to mutations in the tumor suppressor genes PTCH1, SMO and SUFU, or amplifications of GLI2 or MYCN [26,84,86]. TP53 mutations can be found in about 20% of all patients with SHH medulloblastoma and define a “very high risk” group of patients with poor outcome [61,101]. About 20% of patients with SHH tumors present with metastases at time of diagnosis.
Group 3
This subgroup represents about 25% of all medulloblastomas and affects almost exclusively infants and children. A male predominance is typical for this highly aggressive subgroup [27]. A commonly overexpressed pathway has not been identified, however, MYC amplification and isochromosome 17q are frequently observed alterations in these tumors [76,90,99]. In addition, amplification of OTX2, mutation of SMARCA4 and enhancer activation of GFI1 and GFI1B are recurrent genetic alterations [47,77]. Patients with group 3 tumors have the worst outcome and present with leptomeningeal dissemination at time of diagnosis in 40–45% of cases.
Group 4
These medulloblastomas affect patients of all age groups and account for approximately 35% of all medulloblastomas [27]. Although this subgroup is the most common, the underlying pathogenesis is poorly understood and the cells of origin have not been identified. Isochromosome 17q can be found in almost all group 4 tumors, however, there is no association with poor outcome in contrast to that described for group 3 medulloblastomas [76]. Mutation of KDM6A, amplification of MYCN and CDK6, loss of chromosome X in females and duplications of SNCAIP are also frequently detected cytogenetic alterations in this subgroup [44,49,77,79,85]. Despite the frequent presence of metastases at diagnosis, the overall outcome of patients with group 4 medulloblastoma is intermediate.
RISK STRATIFICATION
Traditionally, medulloblastoma patients have been classified into two risk groups, “average risk” and “high risk”, using three clinical criteria : age at diagnosis, presence or absence of leptomeningeal dissemination (based on magnetic resonance imaging and cerebrospinal fluid [CSF] analysis), and extent of residual tumor after resection. “Average risk” patients are older than 3 years of age and present with non-metastatic disease (no macroscopic metastasis on imaging scans and no microscopic tumor cells found in CSF) and a residual tumor size <1.5 cm2. The presence of metastases and/or a postoperative tumor size >1.5 cm2 defines “high risk” disease [46]. Infants (<3 years) are generally considered high risk and are treated using radiation-sparing protocols to reduce neurocognitive side effects.
During a consensus meeting in Heidelberg in 2015, a new risk stratification protocol based on molecular and prognostic criteria was proposed for patients between 3 and 17 years of age [60,61]. The refined classification has four risk groups, mainly defined by outcome, and takes disease heterogeneity and molecular subgroup information into account. The protocol defines patients as “very high risk” (<50% survival), “high risk” (50–75% survival), “standard risk” (75–90% survival), and “low risk” (>90% survival) [61]. Patients with metastatic group 3 medulloblastoma as well as patients with TP53 mutated SHH tumors have a poor prognosis and should be considered very high risk [58,65,80]. High risk patients are patients with metastatic or MYCN amplified SHH tumors as well as group 4 medulloblastoma patients with leptomeningeal dissemination [28,56,61]. Patients with non-MYCN amplified, non TP53 -mutated SHH medulloblastoma, non-MYC amplified group 3 tumors and group 4 tumors without chromosome 11 loss are considered standard risk [61]. Low risk are non-metastatic WNT patients as well as patients with non-metastatic group 4 tumors and whole chromosome 11 loss (Fig. 2) [61].
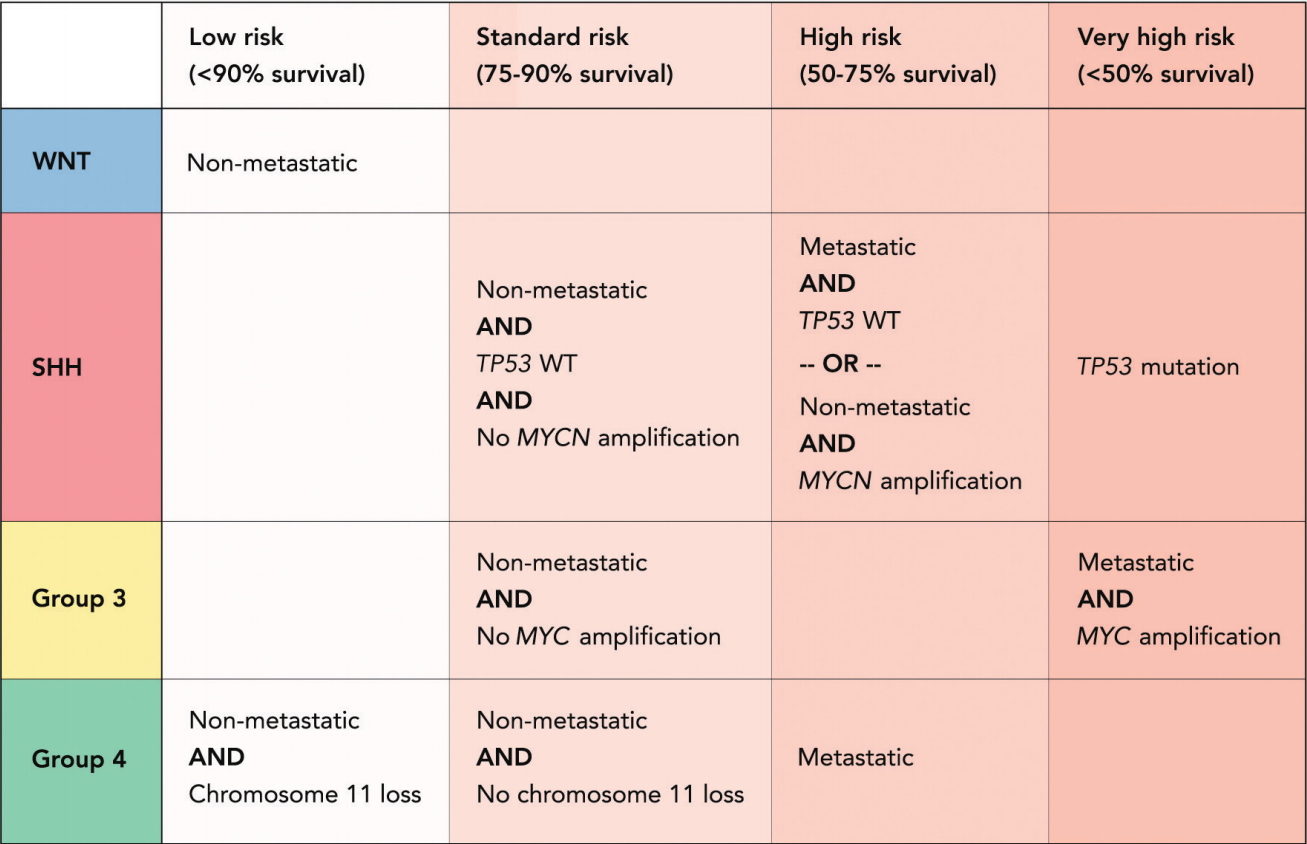
The new patient risk stratification allows for evaluation of treatment de-escalation for patients with favorable outcomes and improves the ability to identify and test new rational, targeted therapies in patients in “high risk” and “very high risk” groups [61].
CURRENT THERAPIES
The current treatment protocols are largely based on the traditional risk stratification and the age of the patient at the time of diagnosis [64]. Patients initially undergo tumor resection at the time of diagnosis, regardless of risk group [29,64]. Recent re-analysis of the prognostic value of extent of resection with subgroup taken into account, demonstrates that there is no benefit for gross total resection over subtotal resection in overall survival for patients, regardless of subgroup, and no overall survival or progression-free survival benefit for patients who underwent near-total versus gross-total resection [88]. Thus, maximal safe surgical resection represents the standard of care for medulloblastoma and there is no apparent clinical benefit of surgical removal of small volume residual disease that carries a high risk of neurological morbidity [88].
Subsequently, “average risk” patients over the age of 3–5 (age cutoffs vary depending on the cooperative group performing the clinical trials) are treated with 23.4 Gy craniospinal irradiation with a boost of 55 Gy to the tumor bed in the posterior fossa and adjuvant cytotoxic chemotherapy [30]. “High risk” patients undergo craniospinal radiation using a dose of 36–39 Gy, a boost of 55 Gy to the tumor bed, and adjuvant chemotherapy [21]. Typical chemotherapy regimens consist of cisplatin/carboplatin-vincristine-cyclophosphamide combinations.
Infants below the age of 3–5 years are currently treated with radiation sparing approaches because of the devastating neuro-cognitive side-effects of craniospinal radiation on the developing nervous system [7,15,17,21,71]. Various chemotherapy regimens have been studied, such as vincristine, cyclophosphamide, etoposide, and cisplatin followed by autologous hematopoietic cell rescue (CCG-99703) and methotrexate (intravenous and intraventricular), vincristine, cyclophosphamide, and carboplatin (HIT-SKK’92) [7,71]. Survival rates in patients with non-desmoplastic histology and macroscopic metastatic disease at the time of diagnosis in this age-group continue to have dismal outcomes [64].
BIOLOGICALLY INFORMED TREATMENT STRATEGIES
The understanding of the heterogeneity that exists within tumors that are broadly classified as medulloblastoma is allowing molecularly stratified trials to be carried out based on both molecular subgroups and an improved understanding of risk stratification [49,61]. The advanced understanding of key molecular alterations within different subgroups (and subtypes) of medulloblastoma provides a basis for the development of risk-adapted treatment protocols and novel targeted therapies specific to molecular events within a particular patient’s tumor [12,21,64,91]. Numerous pre-clinical and clinical trials are underway to develop and test small-molecular inhibitors, antibody-based therapies, and immunotherapies that exploit molecular vulnerabilities in these tumors [2,10,11,21,33].
WNT subgroup
A key strategy being introduced in several active phase II and phase III studies is de-escalation of first-line treatments in lowrisk (non-metastatic) WNT medulloblastoma [21,64]. These trials are designed to reduce (SJMB12, PNET 5 MB) or eliminate craniospinal irradiation (NCT02212574) and implement reduced dose regimens of chemotherapy (PNET 5 MB). Given the excellent overall survival historically noted in these patients (>90%), these studies aim to reduce treatment-related morbidity in these patients with biologically favorable outcomes [19,59,61].
SHH subgroup
Numerous preclinical studies identified hedgehog signaling pathway activation in medulloblastoma and demonstrated evidence of in vitro efficacy of hedgehog pathway inhibitors in medulloblastoma [4,68,81,82]. Vismodegib and sonidegib, competitive antagonists of the smoothened receptor, were among the first targeted therapies to advance to early stage clinical trials in medulloblastoma patients [33,68]. Notably, adult and pediatric medulloblastoma patients treated with vismodegib (PBTC-025B and PBTC-032) exhibited improved progression free-survival in recurrent SHH medulloblastoma, but not non-SHH medulloblastoma [68]. Response to smoothened (SMO) inhibition is highly dependent on the presence of Hedgehog pathway alterations downstream of SMO, including SUFU negative regulator of hedgehog signaling (SUFU) mutations and GLI family zinc finger 2 (GLI2) or MYCN amplifications, which infer resistance to SMO inhibitors [26,51]. SMO inhibition monotherapy has been associated with selection of treatment-resistant subclones, via novel mutations affecting SMO inhibition or upregulation of alternate survival pathways, suggesting that SMO inhibitors will need to be accompanied by additional agents to achieve a durable treatment response [5,69,97].
Alternative agents targeting downstream components of the Hedgehog signaling pathway include arsenic trioxide and itraconazole, inhibitors of the GLI transcription factor, which may be effective in the subset of patients with hedgehog activation independent of SHH-PTCH1-SMO [25,70,83].
Further pre-clinical work has identified additional pathways that may represent targetable vulnerabilities in this subgroup [11,35,36]. For example, in a functional genomic mouse model of SHH medulloblastoma, metastatic populations were enriched for clones with PI3K pathway insertions, suggesting this pathway may be an essential pathway for SHH medulloblastoma metastasis [41,78,96]. Recurrent TP53 mutations are observed in a subtype of SHH medulloblastomas and may represent another targetable pathway to overcome the radiation resistance associated with these mutations [80,100]. Further pre-clinical work is necessary to clarify the ideal candidate therapies and drug combinations to be tested in the next generation of clinical trials.
Group 3 and group 4
Group 3 and 4 tumors currently lack specific targeted therapies in existing clinical trials. However, trial SJMB12 contains a treatment arm for high risk patients, defined as those with metastases at diagnosis, incomplete resection, and/or MYC or MYCN amplified, which will be treated with an additional, novel combination of cytotoxic chemotherapy agents (pemetrexed and gemcitabine) [21]. However, no current clinical trials exclusively enrolling medulloblastoma patients are investigating targeted therapies in these subgroups. Patients with group 3 medulloblastoma currently have the worst prognosis and need to be prioritized for novel treatments [10].
Group 3 and group 4 tumors appear to demonstrate heterogeneity in terms of activated signaling pathways, with MYC amplification being the most common cytogenetic alteration in group 3 tumors [53,92]. Promising pre-clinical agents in group 3 MYC-driven medulloblastoma include combination treatment using PI3K and histone deacetylase inhibitors and BET-bromodomain inhibitors [3,53]. Epigenetic alterations are common in these subgroups, meaning epigenetic targeting may be a promising area for further preclinical investigation [1,9,22,33,48].
METASTATIC MEDULLOBLASTOMA
Medulloblastoma typically metastasizes to the leptomeninges and disseminated leptomeningeal disease represents a formidable treatment challenge. The incidence of metastasis in medulloblastoma at diagnosis is approximately 40% across all subgroups, although each subgroup varies in terms of frequency at diagnosis (Fig. 1) [96,98]. The presence of metastatic disease at the time of diagnosis is a poor prognostic sign in non-WNT subgroups of medulloblastoma [61].
Given the near-universal treatment failure encountered in the setting of relapsed metastatic disease the treatment of the metastatic compartment is now a key focus of investigation [64]. An essential discovery has been that medulloblastoma undergoes significant clonal selection and evolution during the course of the disease, meaning that tumor cells from the metastatic compartment harbor unique genetic and epigenetic alterations not present in the primary tumor [37,93]. Evidence for this includes integrated genomic profiling (copy number, DNA methylation, and whole exome sequencing) of matched tumors from primary and metastatic tumors from human patients and supportive data from a murine transposon-driven SHH-medulloblastoma model, in which common transposon insertion sites were significantly different between the primary and metastatic tumors [37,96]. Thus, metastatic medulloblastoma cells are dependent on molecular pathways for survival in the metastatic niche that are different from the primary tumor, and will likely require specific targeted treatments [37]. Pre-clinical work in this area should focus on identifying mechanisms of medulloblastoma metastasis, while future clinical trials need to account for the unique molecular profile of metastatic medulloblastoma; biopsy of the metastatic compartment to confirm the presence of a treatment target may be necessary to optimize patient selection for experimental therapies.
RECURRENT MEDULLOBLASTOMA
Recurrent medulloblastoma remains extremely refractory to existing therapies, with response rates to various treatment approaches (repeat surgery, re-irradiation, additional chemotherapy regimens, and targeted therapies) at relapse under 10% [13,64,72]. Recent studies have provided significant advances in our understanding of clonal selection events in medulloblastoma from the time of initial diagnosis to recurrence, which is important for biologically informed design of future clinical trials [38].
Tumors of each subgroup have a predilection for specific patterns of spatial and temporal recurrence [37,38,62]. WNT tumors recur in both the primary tumor site and the metastatic compartment, although recurrence rates in this subgroup are low [62]. SHH tumors have a predilection for local recurrence, while group 3 and 4 tumors typically recur with metastatic dissemination [62]. Functional genomic mouse models combined with sequencing of human primary and recurrent samples have demonstrated that the dominant clone at recurrence is present as a minor clone in the primary tumor, and that treatment pressures induce clonal selection and evolution, and lead to the acquisition of novel somatic mutations not present in the primary tumor [38,96]. Design of future clinical trials for recurrent disease should recognize this process and base decisions on resampling and profiling of the recurrent tumor, for proper patient selection for novel therapies.
FUTURE DIRECTIONS
A refined understanding of the molecular underpinnings of medulloblastoma offer significant promise to improve survival of patients with medulloblastoma, and reduce treatment related adverse events, particularly in low risk subgroups. Current work is focusing on defining the heterogeneity within tumor subgroups, this has been most recently demonstrated by multiple groups proposing a refined classification system that identifies new subtypes within each subgroup [6,42,74). These subtypes can provide further clarity in terms of recurrent molecular alterations and could aid in improved selection of patients for targeted therapies, however, there is currently no defined consensus definition of medulloblastoma subtypes.
Significant challenges remain in translating these findings to realize clinical benefit [18]. Accrual of sufficient patients for a clinical trial in medulloblastoma requires multicenter collaboration, and this challenge will be intensified as patients with particular genetic alterations are recruited for trials with targeted therapies [94]. International collaboration will be necessary to determine the ideal design and goals of future clinical trials, to maximize the translation of advances in the molecular understanding of this cancer into improved survival and quality of life for medulloblastoma patients. Neurosurgeons have an important role to play in these endeavors, and given the spatial heterogeneity and evolution of medulloblastoma from the time of treatment to recurrence, biopsies of metastatic and recurrent tumors will likely be necessary to effectively match patients to an optimal targeted therapy.
Notes
CONFLICTS OF INTEREST No potential conflict of interest relevant to this article was reported.
INFORMED CONSENTThis type of study does not require informed consent.
Acknowledgements
The authors would like to thank Stacey Krumholtz, Medical Illustrator at The Hospital for Sick Children, Toronto, for the design of the tables and Dr. Craig Daniels Ph.D. for editing the manuscript.