Memantine Induces NMDAR1-Mediated Autophagic Cell Death in Malignant Glioma Cells
Article information
Abstract
Objective
Autophagy is one of the key responses of cells to programmed cell death. Memantine, an approved anti-dementia drug, has an antiproliferative effect on cancer cells but the mechanism is poorly understood. The aim of the present study was to test the possibility of induction of autophagic cell death by memantine in glioma cell lines.
Methods
Glioma cell lines (T-98 G and U-251 MG) were used for this study.
Results
The antiproliferative effect of memantine was shown on T-98 G cells, which expressed N-methyl-D-aspartate 1 receptor (NMDAR1). Memantine increased the autophagic-related proteins as the conversion ratio of light chain protein 3-II (LC3-II)-/LC3-I and the expression of beclin-1. Memantine also increased formation of autophagic vacuoles observed under a transmission electron microscope. Transfection of small interfering RNA (siRNA) to knock down NMDAR1 in the glioma cells induced resistance to memantine and decreased the LC3-II/LC3-I ratio in T-98 G cells.
Conclusion
Our study demonstrates that in glioma cells, memantine inhibits proliferation and induces autophagy mediated by NMDAR1.
INTRODUCTION
Malignant gliomas are the most common primary tumors of the central nervous system (CNS) and show poor prognosis in spite of multimodality treatments, including extensive tumor resection, radiation therapy and chemotherapy26). Recently, many clinical trials of targeted antiglioma agents were conducted, but they failed to elicit a therapeutic response in malignant gliomas2). The possible reasons for poor outcomes in malignant gliomas are the microenvironment and characteristics of glioma cells. The unique environments of the CNS, for example, protection by the blood-brain barrier and blood-tumor barrier, interfere with the passage of therapeutic agents from the blood to the brain tumor. In addition, the highly proliferative and invasive behavior of glioma cells limits the benefits of surgical resection15).
Glutamate, an excitatory neurotransmitter released by certain neuronal cells in the CNS, is also secreted by glioma spontaneously and facilitates invasion by the tumor cells24). Glutamate also has an autocrine activity inducing the proliferation of glioma cells27). Glioma cells express multiple glutamate receptor subtypes including amino-3-hydroxy-5-methyl-4-isoaxazolepropionate acid receptor (AMPAR) and N-methyl-D-aspartate receptor (NMDAR)7). In glioma cells, AMPAR is believed to promote cellular proliferation and survival, via activation of the PI3K/AKT pathway, EGFR expression and the MAPK pathway8,10,23). NMDAR antagonists also have the potential for inhibiting tumor proliferation1,17,22,25). Dizocilpine, an NMDAR1 antagonist, inhibits proliferation of lung cancer cell lines by inhibiting the extracellular signal-regulated kinase 1/2 pathway25). At present, the NMDAR1-mediated antiproliferative pathway is poorly understood, particularly in glioma.
NMDARs consist of 3 subtypes: NMDAR1, NMDAR2, and NMDAR3. NMDAR2 has 4 subunits (A–D), and NMDAR3 has 2 subunits (A and B). NMDAR1 and NMDAR2 are dependent on glutamate11). NMDAR2A and NMDAR2B perform different functions in the CNS. The NMDAR2A subunit is primarily found in the synapses and is required for glutamate-mediated neuronal survival. However, the NMDAR2B subunit is primarily found in the extrasynaptic sites and is involved in glutamate-mediated neuronal death6,14). An association between NMDAR2B signaling and autophagic cell death was reported in traumatic brain injury (TBI)5). TBI induces redistribution of the NMDAR2B multi-protein signaling complex and upregulates expression of the autophagic protein beclin-1. Ro25-6981, an NMDAR2B antagonist, influences autophagic cell death in TBI.
Memantine, an NMDAR1 antagonist, is clinically effective in Alzheimer’s dementia and Parkinson’s disease29). Memantine has an antiproliferative effect on tumor cells1,17,22,25), but its effect on autophagy has yet to be elucidated. The aim of this study was to test whether memantine can cause autophagic cell death in glioma cells.
MATERIALS AND METHODS
Cell culture
T-98 G and U-251 MG cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). T-98 G cells were grown in Eagle’s minimum essential medium and U-251 MG cells in Dulbecco’s Modified Eagle’s Medium. All media were supplemented with 2 mM/L L-glutamine, 100 units/mL penicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum (FBS) purchased from Invitrogen (Carlsbad, CA, USA). Cells were incubated at 37°C in a humidified atmosphere containing 5% CO2.
Growth-inhibitory effects of memantine on glioma cells
Inhibition of growth of the glioma cell lines by memantine was evaluated to identify from the concentration required for 50% growth inhibition (IC50) in comparison with untreated controls. Cells were seeded in a 96-well plate at a density of 104/well in a medium containing 10% FBS and were incubated for 48 h. The cells were subsequently washed twice with the medium and incubated further with fresh medium (control) or with the medium containing 10–600 μM of memantine. After exposure to memantine for 48 h, the cells were harvested by trypsinization and viability of the cell population was determined using the CCK-8 assay kit (Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions. The experiments were repeated thrice at each concentration.
Western blot analysis
Antibodies were obtained from commercial sources: anti-NMDAR1, anti-NMDAR2B, and anti-LC3A/B; anti-APG5 and anti-ULK1 (Abcam, Cambridge, UK); anti-beclin1, anti-AMP-activated protein kinase (AMPK) α, and β-actin (Santa Cruz Biotechnology Inc., Dallas, TX, USA).
For the western blot analysis, the cells were rinsed with phosphate-buffered saline (PBS) and subsequently lysed for 30 min on ice in RIPA-B buffer (0.5% Nonidet P-40, 20 mM Tris-HCl, pH 8.0, 50 mM NaCl, 50 mM NaF, 100 μM Na3VO4, 1 mM DTT, and 50 μg/mL PMSF). The insoluble material was removed by centrifugation at 12000 rpm for 20 min. Next, the supernatant was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis. The blots were blocked in PBS with 5% skim milk and 0.05% Tween 20, incubated with the diluted primary antibodies against NMDAR1, NMDAR2B, or LC3A/B (1: 1000; Cell Signaling Technology, Denvers, MA, USA); APG5 or ULK1 (1: 1000; Abcam, Cambridge, UK); beclin-1 or AMPKα (1: 200; Santa Cruz Biotechnology Inc., Dallas, TX, USA), and followed by incubation with a horseradish peroxidase-conjugated secondary antibodies (1: 2500; Santa Cruz). β-actin (Santa Cruz) was used as a control. The blots were assayed using an enhanced chemiluminescence detection system (Image Quant LAS 4000mini, Pittsburgh, PA, USA).
Reverse transcription polymerase chain reaction (RT-PCR)
Total RNA was isolated from the glioma cell lines using RNAiso Plus (Takara, Ostu, Japan) according to the manufacturer’s instructions. We synthesized cDNA using 3 μg of total RNA and an oligo (dT) primer and PrimeScript reverse transcriptase for reverse transcription PCR (Takara). PCR amplifications consisted of a total of 37 cycles of denaturation at 94°C for 45 s, annealing at 62°C for 45 s, extension at 72°C for 1 min with the initial denaturation at 94°C for 5 min, and final extension at 72°C for 5 min. NMDAR1 primers used in the PCR analysis included forward primer 1, 5′-ATC TAC TCG GAC AAG AGC ATC C-3′ and reverse primer 1, 5′-AGC TCT TTC GCC TCC ATC AG-3′. NMDAR2B primers included forward primer 1, 5′-TCA AGG ATG CCC ACG AGA AAG-3′ and reverse primer 1, 5′-GTG GCT TCT TCC TTG TGA CAG-3′.
Transfection with small interfering RNA (siRNA)
NMDAR1 siRNA was obtained from Genolution (Seoul, Korea). To transfect tumor cells with siRNA, cells (104/well) were seeded in 96-well plates and Lipofectamine 2000 (Invitrogen) with siRNA were added according to the manufacturer’s instructions. The cells transfected with scrambled siRNA (Genolution) were used as controls.
Transmission electron microscopy (TEM)
Cell samples were fixed overnight in 4% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M phosphate buffer. After washing in 0.1 M phosphate buffer, the samples were postfixed with 1% osmium tetroxide in the same buffer for 1 h. Following which, the cell samples were dehydrated using a graded series of ethyl alcohol (50, 70, 90, and 100%) and pure acetone. The specimens were embedded in Epon 812 and the polymerization was performed at 60°C for 3 days. Ultrathin sections (60–70 nm) were made on an ultramicrotome (Leica Ultracut UCT, Wetzlar, Germany) and were examined under a TEM (JEM 1010, Tokyo, Japan) operating at 60 kV. The TEM images were acquired using a CCD camera (SC1000, Pleasanton, CA, USA).
Statistical analysis
Statistical analysis was performed using Student’s t-test. Data were calculated as mean±standard errors (SE). Differences were considered statistically significant at p<0.05.
RESULTS
Antitumor effects of memantine on human malignant glioma cells
To determine the antiproliferative effects of memantine on malignant glioma cells, we incubated malignant glioma cell lines with 0–600 μM of memantine for 48 hr and assessed the percentage of viable cells. T-98 G cells showed growth inhibition by memantine in a dose-dependent manner. The IC50 value of memantine in T-98 G cells was 400 μM. The IC50 value in U-251 MG cells was not identified (Fig. 1). Based on these results, memantine had an antiproliferative effect on T-98 G cells.
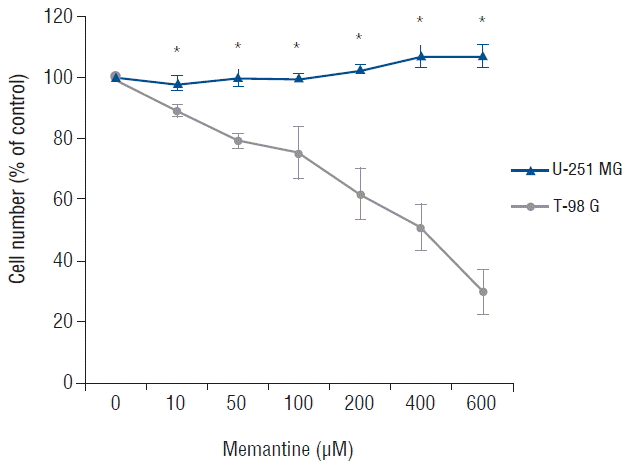
Memantine-induced cytotoxicity in glioma cell lines. After incubation with memantine (0–600 μM), malignant glioma cells were grown for 48 h starting at a density of 104/well. The number of viable cells was counted and expressed as a percentage of the untreated control. The results are representative of 3 independent experiments. *p<0.05, Student’s t-test.
Expression of NMDAR in human malignant glioma cells
Because memantine acts as an antagonist for NMDAR in competition with glutamate, we analyzed expression of NMDAR1 and NMDAR2B in glioma cell lines. On a western blot, the NMDAR1 protein was identified in T-98 G cells, and not in U-251 MG cells. In contrast, NMDAR2B proteins were identified in both U-251 MG cells and T-98 G cells (Fig. 2A). On RT-PCR, NMDAR1 mRNA was detectable only in T-98 G cells, and NMDAR2B mRNA was present in both glioma cells (Fig. 2B). These results suggested that the sensitivity of T-98 G cells to memantine may be related to the expression of NMDAR1.

Expression levels of NMDAR1 and NMDAR2B in U-251 MG and T-98 G cells. The cell lines were analyzed using western blotting (A) and RT-PCR (B). NMDAR1 was present only in T-98 G cells, but NMDAR2B was detectable in both glioma cell lines. β-actin and GAPDH were used as internal controls. NMDAR: N-methyl-D-aspartate receptor, RT-PCR: reverse transcription polymerase chain reaction, GAPDH: glyceraldehyde-3-phosphate dehydrogenase.
Induction of the autophagic process in T-98 G cells by memantine
Autophagy is a type of programmed cell death characterized by double-membrane autophagosome (AP) involving several autophagy-related proteins. The conversion of light chain protein 3-I (LC3-I) to LC3-II is an essential process for AP formation13). We examined the expression of LC3-1 and LC3-II using western blotting after incubation of T-98 G cells with 0 and 400 μM of memantine. The immunoblots were analyzed using densitometry, which quantified the amount of protein staining, and calculated relative amounts compared with the internal control, β-actin. After the incubation with memantine (400 μM) for 48 h, LC3-II was expressed and the LC3-II- /LC3-I ratio was significantly increased in T-98 G cells (Fig. 3A). In addition, beclin-1, another autophagy-related protein, showed a significant increase of expression. Other autophagy-related proteins, AMPKα, APG5L, and ULK-1, were not unregulated (Fig. 3B).
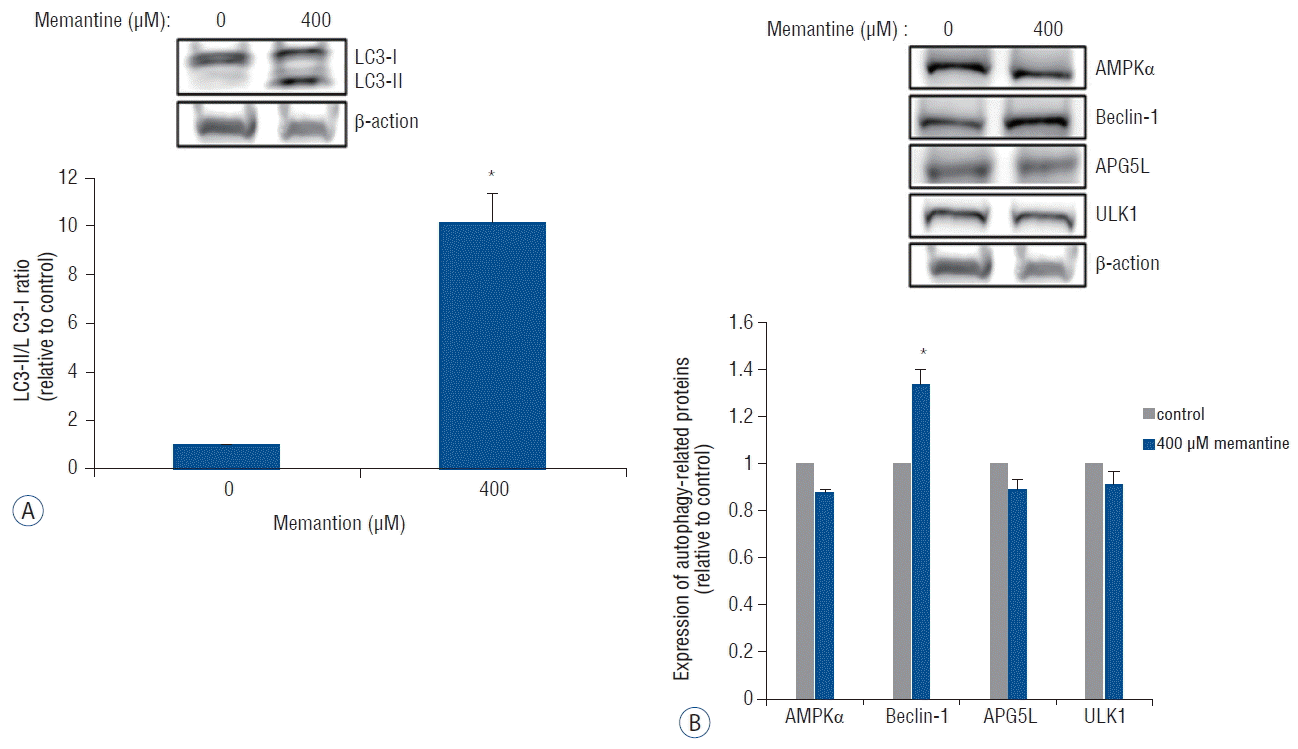
Memantine upregulated LC3-II and beclin-1 in T-98 G cells. A: Protein levels of LC3-I and LC3-II were analyzed in T-98 G cells treated with 0 and 400 μM memantine for 48 h. The graph shows the results of densitometric quantitation of western blots, presented as the LC3-II/LC3-I ratio relative to the control (0 μM of memantine). LC3-II was significantly increased by 400 μM memantine. B: Protein expression levels of AMPKα, beclin-1, APG5L, and ULK1 were evaluated using western blotting after incubation with memantine (0 and 400 μM) for 48 hr in T-98 G cells. The results of densitometric quantitation of immunoblots are presented as the ratio relative to the control (0 μM of memantine). Beclin-1 was significantly upregulated after the treatment with 400 μM of memantine. *p<0.05, Student’s t-test. AMPK: AMP-activated protein kinase.
Identification of autophagic using by TEM in T-98 G cells
The ultrastructural features of T-98 G cells after the memantine treatment were evaluated using TEM. The media-treated control cells exhibited intact cytoskeletal architectures and had normal cell organelles, such as mitochondria, ribosomes, and endoplasmic reticulum (Fig. 4A). After incubation with 400 μM memantine for 48 h, T-98 G cells preserved the cytoskeleton but developed the lamellar-structured autophagic vacuoles in the cytoplasm (Fig. 4B). Based on these results, in T-98 G cells, memantine most probably induced autophagic cell death.

Memantine induced autophagic ultrastructural features in T-98 G cells according to TEM. A: The control cells showed the normal cytoplasm including mitochondria, ribosomes, and endoplasmic reticulum. B: After incubation with 400 μM memantine for 48 h, the cells developed autophagic vacuoles with lamellar structure in the cytoplasm. TEM: transmission electron microscopy.
A knockdown of NMDAR1 by siRNA interferes with autophagic cell death of T-98 G cells
To confirm the functional significance of NMDAR1, we tested whether NMDAR1-silencing would reverse the antiproliferative effect of memantine on T-98 G cells. SiRNA oligos were designed complementary to the human NMDAR1 mRNA. Forty-eight hours after the transfection of anti-NMDAR1 siRNA into T-98 G cells, a reduction of NMDAR1 expression was confirmed using western blot analysis (Fig. 5A). The differences in cell viability between NMDAR1 knockdown and control T-98 G cells were analyzed using incubation with 0–600 μM of memantine with a subsequent CCK-8 assay. A significant increase in cell viability of NMDAR1 knockdown cells was detected at the memantine doses of 400 and 600 μM (Fig. 5B). The expression of LC3-II and LC3-I was evaluated after incubation of NMDAR1 knockdown and control T-98 G cells with 400 μM memantine. The LC3-II/LC3-I ratio was decreased in NMDAR1 knockdown cells (Fig. 5C). These results suggested that NMDAR1 was necessary for autophagic cell death of glioma cells.

A knockdown of NMDAR1 using siRNA transfection in T-98 G cells. T-98 cells incubated in EMEM was used as the control value. A: After the transfection with anti-NMDAR1 siRNA (10 nM) for 48 hr, the expression of NMDAR1 was evaluated using western bloting. B: Cell viability of control and siRNA-transfected T-98 G cells was analyzed after incubation with various concentration of memantine (0–600 μM). Significant differences in cell viability was observed at 400, and 600 μM of memantine. C: Protein level of LC3-I and LC3-II was evaluated in control and NMDAR1 knockdown cells using western blotting after incubation with 400 μM memantine. The results of densitometric quantitation of immunoblots were represented as the LC3-II/LC3-I ratio relative to control cells. *p<0.05, Student’s t-test. NMDAR: N-methyl-D-aspartate receptor, siRNA: small interfering RNA, EMEM: Eagle’s minimum essential medium.
DISCUSSION
Glutamate is the main excitatory neurotransmitter in the CNS and is released from active neurons through calcium-dependent fusion of synaptic vesicles7). Recent studies have shown that glioma cells release glutamate spontaneously. A monolayer of glioma cells maintained in a culture flask for 12 h achieves glutamate concentrations exceeding 500 μM30). Microdialysis in RG2 glioma cells implanted into the cortex of a rat shows that the peritumoral region undergoes 4-fold elevation of the glutamate concentration compared with either the tumor or healthy-brain tissue3). Some studies examined the glutamate concentration using non-invasive techniques such as magnetic resonance spectroscopy (MRS). The concentration of glutamate was shown to be elevated in the peritumoral area in patients with a brain tumor9,19). The glutamate released from glioma cells has several effects on the surrounding neuronal, glial, and tumoral cells. The excessive release of glutamate from glioma causes peritumoral excitotoxicity with neuronal cell death as well as tumor expansion via stimulation of the glutamate receptor NMDAR7). Excitotoxicity also manifests itself as hyperexcitability in the peritumoral tissue and results in seizures16,24). In addition, glutamate stimulates tumor cell motility and invasion via another glutamate receptor, AMPAR18). Research shows that glutamate promotes glioma cell proliferation and survival27). Several studies reported that a glutamate receptor antagonist inhibits tumor growth, whereas a glutamate scavenger shows positive results in survival of animals with glioma21,22). NMDAR antagonists, such as memantine, MK-801 (dizocilpine), ifenprodil, Ro25-6981, and AP5, have an antiproliferative effect on cancer cells1,17,22,25,28). Our study also showed that memantine has an antiproliferative effect on human glioma cell lines. Nevertheless, the sensitivity to memantine is different among glioma cell lines. T-98 G cells are sensitive but U-251 MG cells are resistant to memantine. To identify a possible reason for the difference in sensitivity, we analyzed the expression of NMDAR in those cell lines because memantine is an NMDAR1 antagonist. The expressions of NMDAR1 and NMDAR2 was evaluated in glioma cell lines using both western blotting and RT-PCR. Only T-98 G cells express NMDAR1 and this observation correlates with the sensitivity to memantine.
NMDAR plays different roles in the CNS, depending on the subtypes6,14). Excessive activation of NMDAR2B is believed to be a major mechanism responsible for cell death associated with several neurological diseases such as brain trauma and some several neurodegenerative diseases20). Selective Ca2+ influx through NMDAR2B is associated with neuronal cell death, and NMDAR2B signaling efficiency is dependent on receptor localization within lipid rafts of the plasma membrane4,20). Bigford et al. reported that moderate TBI induces recruitment of NMDAR2B and translocation of beclin-1 out of the membrane microdomains5). Beclin-1 regulates the autophagic pathway by forming a complex with type III phosphatidylinositiol-3-kinase, responsible for autophagic vesicle nucleation12). The release of beclin-1 from the complex in response to excessive stimulation of NMDAR2B induces activation of autophagy in TBI5). In our study, T-98 G cells expressed NMDAR1 but underwent autophagic cell death after incubation with NMDAR1 antagonist memantine. The conversion ratio of LC3-I to LC3-II and expression of beclin-1 were elevated in T-98 G cells as a result of memantine action. Ultrastructural analysis of T-98 G cells showed autophagic vacuoles after incubation of the cells with memantine. The role of NMDAR1 in autophagy was confirmed by the transfection of NMDAR1 siRNA into T-98 G cells. The siRNA transfection knocked down NMDAR1 protein expression successfully, and the antiproliferative effect of memantine was attenuated, whereas the LC3-II/LC3-I ratio was decreased in T-98 G cells.
CONCLUSION
In conclusion, this study appears to be the first to demonstrate that memantine inhibits proliferation and induces autophagic cell death in glioma cells. We also show that these effects are mediated by NMDAR1. These finding are expected to advance the understanding of molecular mechanisms that underlies the resistance of malignant glioma to existing anticancer modalities.
Acknowledgements
The research was supported by the Clinical Research Laboratory of Incheon St. Mary’s Hospital, The Catholic University of Korea.