Misunderstanding of Foot Drop in a Patient with Charcot-Marie-Tooth Disease and Lumbar Disk Herniation
Article information
Abstract
We report the case of 57-year-old woman diagnosed with Charcot-Marie-Tooth (CMT) disease and lumbar disk herniation (LDH). She had left leg weakness and foot numbness, foot deformity (muscle atrophy, high arch, and clawed toes). The lumbar spine MRI showed LDH at L4-5. Additionally, electrophysiology results were consistent with chronic peripheral motor-sensory polyneuropathy (axonopathy). In genetic testing, 17p11.2-p12 duplication/deletions characteristic of CMT disease were observed. We confirmed the patient's diagnosis as CMT disease and used conservative treatment.
INTRODUCTION
Generally, lumbar disk herniation (LDH) with foot drop is an indication for early decompressive surgery7). However, even for cases of painless foot drop or foot drop with numbness, examinations are necessary to distinguish them from other diagnoses including peroneal nerve palsy, sciatic nerve mononeuropathy, lumbosacral plexopathy, polyneuropathy, and severe L5 radiculopathy1,5). We report here an unusual case of Charcot-Marie-Tooth (CMT) disease initially presenting with unilateral foot drop with foot numbness.
CASE REPORT
A 57-year-old woman, presenting with left leg weakness and foot numbness, visited our hospital for surgery after being diagnosed with LDH at a local hospital. The patient was treated with neuroplasty twice at the local hospital, but did not show symptom improvements. Microdiscectomy was recommended.
On neurological examination, left ankle dorsiflexion weakness (grade III) and knee extension weakness (grade IV) with foot numbness were observed. Additionally, we found foot deformity (muscle atrophy, high arch, and clawed toes) (Fig. 1). MRI of the lumbar spine showed LDH at L4-5 (Fig. 2). Her neurological status and symptoms were not correlated with L5 radiculopathy. To obtain a differential diagnosis, we performed further examinations. To evaluate neuropathy or myopathy, we performed an electrophysiological study. Electrophysiological findings were consistent with chronic peripheral motor-sensory polyneuropathy (axonopathy). The neurologist presumed CMT disease and recommended a genetic test or nerve biopsy for its confirmation. In genetic testing, the 17p11.2-p12 duplication/deletions characteristic of CMT disease were observed. We confirmed the diagnosis of CMT disease in this patient and used conservative treatment.

Foot photography showing muscle atrophy, high arch, and clawed toes.
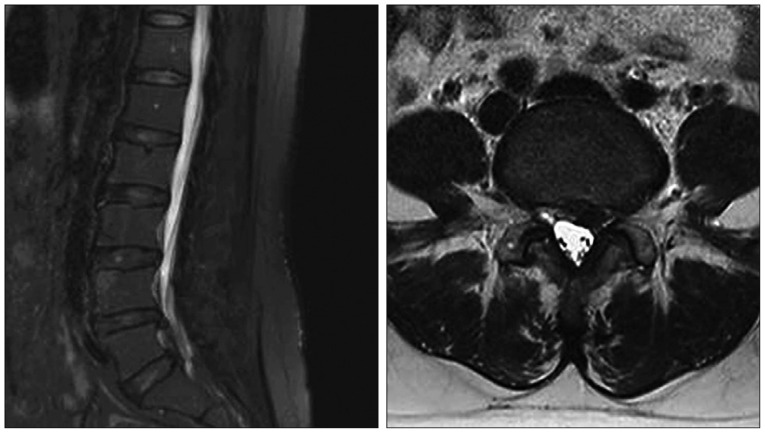
Lumbar MRI showing lumbar disk herniation on the L4-5 left side. The left L5 root is mildly compressed by a herniated disc.
DISCUSSION
CMT disease is a peripheral neuropathy and a subtype of hereditary motor-sensory neuropathies (HMSN)9,10). It is the most common type of inherited polyneuropathy, with a prevalence estimated at up to 40 individuals in every 1000001,9). CMT disease is distinguished from the demyelinating type, HMSN type 1, from axonal HMSN type 26,9,14,15). Both HMSN types are genetically heterogeneous, resulting from various genetic defects3), the most common being duplication or deletion of the peripheral myelin protein gene (PMP-22) gene at chromosome 17, which results in either phenotype1,4,6,9,15).
Clinically, CMT patients often initially present symmetrical distal predominance of limb-muscle wasting, weakness, and sensory loss, especially in the lower extremities9,10,15). Motor symptoms from the feet include pes cavus (or pes planus) (Fig. 1), hammer toes, difficulty in running, walking, twisting of the ankle, tripping, foot drop, steppage gait, wasting, and weakness9,10,13). Hand tremor, muscle cramps, cold feet, and acrocyanosis are other common signs9). Positive sensory symptoms such as paresthesia, numbness, and radiating pain are common, leading to operations based on misdiagnoses9). Deep tendon reflexes are reduced or absent following the same distal to proximal gradient, and some patients have skeletal deformities, including scoliosis9,10,13). Because these symptoms are similar to complaints in patients with spinal myelopathy or radiculopathy, spine surgeons could misdiagnose a patient with CMT disease.
For diagnosis, clinical findings and identification of hereditary patterns could be useful, of which autosomal-dominant inheritance is the most common pattern9,13,14,15). It is important to bear in mind that X-linked Charcot-Marie-Tooth (CMTX1), transmitted as an X-linked dominant trait, is characterized by no male-to-male transmission9,13,15). Useful diagnostic methods may include electromyography and nerve conduction velocity. Electrophysiological studies should be performed to assess the presence, degree, and pattern of nerve-conduction slowing. Electrophysiologically, CMT1 is a uniform, diffuse demyelinating neuropathy with marked slowing of nerve conduction velocities (<38 m/s in upper-limb motor nerves)3,8,9,10,15). Conduction slowing is greater in men than women3,10,13). CMT2, on the other hand, shows less severe conducting slowing and may even exhibit no such slowing at all (>38 m/s) in CMT23,9,14,15). Additionally, molecular tests are necessary to confirm CMT. However, DNA testing needs to take into account the fact that there are numerous genes associated with CMT1,14). In autosomal-dominant or sporadic CMT with electrophysiological evidence of demyelination, CMT1A duplication should be investigated first, followed by consideration of CMTX1 and mutations in gap junction β-1 protein. If CMT2 is diagnosed, molecular tests should be directed toward mitofusin-2 and myelin protein zero9,10,11,13). In addition, nerve biopsy can be useful to confirm CMT. However, we did not perform this test in our case. Nerve biopsy shows characteristic focal hypermyelination involving many internodes that cause segments of thickened myelin resembling links of sausage (tomaculi), hence its original name of tomaculous neuropathy9,15).
Spine surgeons should be aware that painless foot drop or foot drop with nonspecific numbness could be caused by peroneal nerve palsy, sciatic nerve mononeuropathy, lumbosacral plexopathy, polyneuropathy, and severe L5 radiculopathy1,10). These diseases should be excluded expeditiously, because severe cases of LDH could show progressing weakness coupled with a gradual reduction of pain. In our case, the patient had extruded LDH, which confused the cause of the foot drop for physicians at a local hospital. Indeed, elderly or middle-aged patients may have asymptomatic LDH or stenosis2,12); we should therefore be cautious about deciding on spine surgery for patients with a degenerative spine.
CONCLUSION
When we treat patients with painless foot drop or foot drop with numbness, we recommend careful diagnosis. It is necessary to distinguish between various diseases, such as peripheral neuropathy and severe radiculopathy. In addition, CMT should be considered as a cause, although its incidence is rare, because its various symptoms are similar to spinal disease or peripheral neuropathy.
Acknowledgements
This research was supported by Kyungpook National University Research Fund, 2012.