INTRODUCTION
During the last couple of decades, the number of births with spinal open neural tube defects (ONTDs), myelomeningocele (MMC), has rapidly decreased in developed countries, including Korea, because of genetic counseling, prenatal prophylaxis and diagnosis, improved nutrition and sanitation, and the termination of pregnancy. Instead, owing to increased levels of attention among neurosurgeons, pediatricians and urologists and advanced diagnostic tools such as magnetic resonance imaging (MRI) and urodynamic studies, the number of cases with occult spinal dysraphism, mainly lumbosacral lipomatous malformation (LLM), is rapidly increasing. Simultaneously, secondary neurulation, with key words such as the primitive streak, primitive node (Hensen’s node), gastrulation, junctional neurulation, caudal cell mass (CCM) and medullary cord, became one of the central topics of academic interest in this field.
The advancement of knowledge on secondary neurulation, in addition to the development of technology such as intraoperative neurophysiological monitoring (IONM) and new pioneers in this field, has led to dramatic changes in the understanding and management techniques in occult spinal dysraphism seen in the last two decades [62].
In this article, various forms of dysraphic entities and associated anomalous lesions of secondary neurulation are described, mainly focusing on pathoembryogenesis (Table 1). The normal process of secondary neurulation is described in other preceding articles by leading scholars on this topic. Therefore, the details of normal secondary neurulation are not described in this article.
Some ideas suggested in this article were extrapolated from the data of chick embryos. As research on human embryos or fetuses has limitations, research on chick embryos, which are known to have closer similarity to human embryos in terms of secondary neurulation than rodent embryos, is a good alternative. Although the findings in chick embryos may not be directly applied to human embryos, the data still shed some light on human developmental processes. We expect the speculations in this article to provoke further research to obtain deeper insights into secondary neurulation disorders, even though some ideas are conjectural, controversial and contradictory to previous understanding.
Each part of this article was primarily written by one of the coauthors and modified after discussion among the authors : Jeyul Yang (CCM duplication associated with disturbed activity of caudal mesenchymal tissue), Ji Yeoun Lee (hypoplasia or arrest of secondary neurulation associated with disturbed activity of caudal mesenchymal tissue, failed ingression of the primitive streak to the CCM), Kyung Hyun Kim (failed regression spectrum of the medullary cord) and Kyu-Chang Wang (failed junction with the primary neural tube, focal limited dorsal neuro-cutaneous nondisjunction, neuro-mesenchymal adhesion).
FAILED JUNCTION WITH THE PRIMARY NEURAL TUBE
As described in a preceding article, junctional neurulation and junctional neural tube defect (JNTD) are recently introduced terms [13,17]. The details are not provided in this article.
The presence of an overlapping zone between the primary and secondary neural tubes is previously well known [12,58]. In 2016, Eibach et al. [17] reported three cases lacking functional connection between the primary and secondary neural tubes and coined the term ‘JNTD’. In normal embryogenesis, the primary and secondary neural tubes are never structurally separated. The reported cases of Eibach et al. [17], which had nonfunctional band-like structures instead of a functional spinal cord at the junctional zone, must have had localized insults at the place of junctional neurulation. The authors pointed to the loss of function of Prickle-1 as the possible molecular background of the JNTDs, citing the experimental data of Dady et al. [13], although the degree of perturbation was smaller in their human cases. In 2020, Wang et al. [63] described two cases of segmental spinal dysgenesis (SSD) with a review of previously reported cases. Based on the similarity of spinal cord pathology, they insisted that SSD, which was established as a separate entity by Scott et al. [57] in 1988, and the newly introduced JNTD share the same pathoembryogenetic background.
Using a marker study in chick embryos and the extrapolation of the data to human embryos, Dady et al. [13] showed that the junctional zone between the primary and secondary neural tubes ranges from the lower thoracic to upper sacral spinal cord segments in humans. This information solved the enigmas of several dysraphic lesions, such as caudal agenesis, caudal duplication syndrome (CDupS), SSD, and LLM of transitional type, in which the main pathology seemed to occur during secondary neurulation, but the spine and spinal cord of lower thoracic and lumbar segments (previously regarded as products of primary neurulation) may be involved. Knowledge of junctional neurulation will widen the pathoembryogenetic understanding of many lower spinal dysraphic entities.
HYPOPLASIA OR ARREST OF SECONDARY NEURULATION ASSOCIATED WITH DISTURBED ACTIVITY OF CAUDAL MESENCHYMAL TISSUE
The details of caudal agenesis and related multiorgan manifestations are described in a preceding article, and this section will mainly focus on pathoembryogenesis. The wide spectrum and complexity of the associated anomalies can be attributed to the pathoembryogenesis of caudal agenesis [5,14,44]. The obvious involvement of the lumbosacral region places secondary neurulation and the CCM in the core of the pathoembryogenesis of the disease. A striking difference between primary and secondary neurulation is that the CCM is thought not only to give rise to the lumbosacral spinal cord but also to affect the development of surrounding structures such as the urogenital and distal gut [41]. Therefore, an insult to the CCM may result in a complex anomaly involving multiple organs.
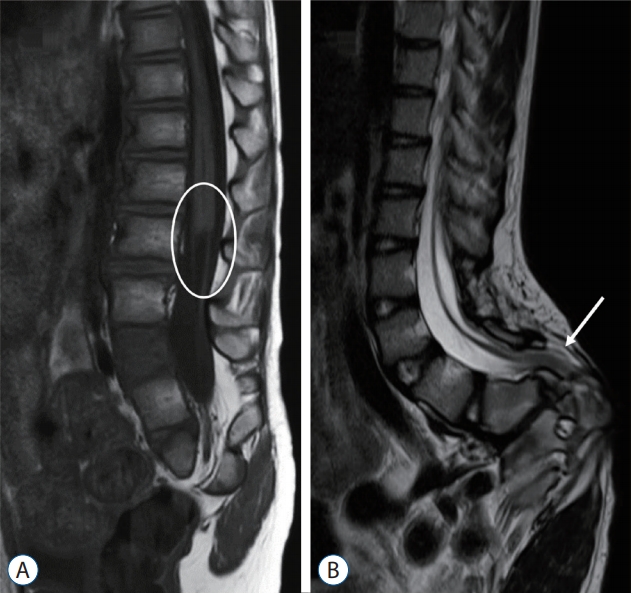
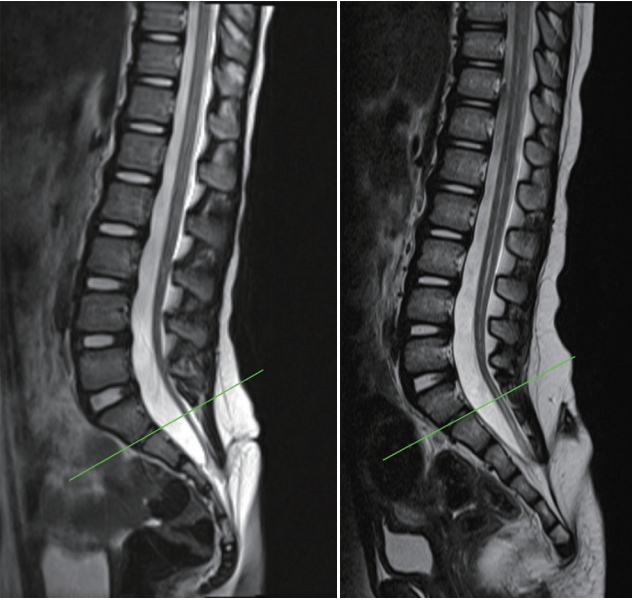
Regarding the spinal cord, insult during the earlier period of secondary neurulation will result in failed formation of the medullary cord. The lower spinal cord including the conus is affected, and the patients typically show a ‘blunt-ended’ conus (Fig. 1A). It is of note that the upper range of the level of the blunt conus can go as far up as the lower thoracic spine [46]. In fact, the level of the blunt type of conus is frequently at the thoracolumbar junction. This is interesting because it exceeds the typical ‘secondary neurulation’ region. The blunt type of conus is speculated to be the result of the ‘failure of the formation of the secondary neural tube’, and its level represents the range of the junction between the primary and secondary neural tubes, as stated in the previous section, ‘Failed junction with the primary neural tube’. Although patients may have various symptoms, including voiding difficulty, lower extremity weakness and atrophy, the majority of them will not benefit from neurosurgical intervention, such as the transection of the filum. In the most severe type of formation failure, not only the spinal cord but also the sacral vertebrae and the urogenital and/or anorectal components may not be formed or are abnormally formed, even with fused lower extremities (the sirenoid or mermaid appearance) [46].
Insult during the later period of secondary neurulation brings about the ‘failure of the regression of the medullary cord’. A thickened and/or fatty filum with a relatively normal looking conus at the normal or lower level is frequently seen (Fig. 1B). The usual manifestation of cord tethering may be found, for which untethering is needed. Caudal agenesis may be associated with more profound types of spinal dysraphism, such as caudal-type LLM or terminal myelocystocele (TMCC), and these may be included in the ‘failure of regression’ group.
It is postulated that insufficient development of the caudal mesenchyme from the CCM may be the main player in the formation of the various types of urogenital/anorectal anomalies. The deficient supply of caudal mesenchyme will result in local defective growth or a lack of enough caudal tissue for effective caudal-ventral push of the caudal mesoderm and cloacal membrane [46]. As the proper development of the caudal mesenchyme composing the walls of the cloaca is important for the formation of the urorectal septum and its approximation to the area of the cloacal membrane, defects in the caudal mesenchyme may lead to various types of fistulous or occluded malformations involving the urogenital/anorectal organs [33]. Cloacal exstrophy may be speculated to be the result of the rupture of the abnormally large cloacal membrane due to extreme deficiency in the ventral push of the caudal mesenchyme [43].
The wide variation in the clinical phenotype of caudal agenesis may be explained by the complex embryology of the CCM and secondary neurulation. Clinicians should be aware of the variability in the presentation and symptoms based on pathoembryogenesis for proper management of patients.
DUPLICATION OF THE CCM ASSOCIATED WITH DISTURBED ACTIVITY OF CAUDAL MESENCHYMAL TISSUE
Since the 18th century, patients manifesting various clinical anomalies of duplicated distal organs have rarely been reported [20]. In 1993, Dominguez et al. [15] reported six patients with complex anomalies of the distal caudal end of the trunk and proposed the term CDupS. The pathoembryogenesis of CDupS remains unclear owing to its rare incidence and limited case reports. A few hypotheses regarding pathoembryogenesis have been proposed. However, due to the absence of a systematic review, weak points of the hypotheses are also present. On the other hand, we noticed that these patients shared common features despite a wide range of clinical manifestations by reviewing the literature on CDupS. Moreover, CDupS cases have common clinical manifestations with caudal agenesis, which involves similar organs but in a different manner. In a situation where it is inevitable to rely on rare case reports, inference based on common clinical findings is the key to explaining pathoembryogenesis.
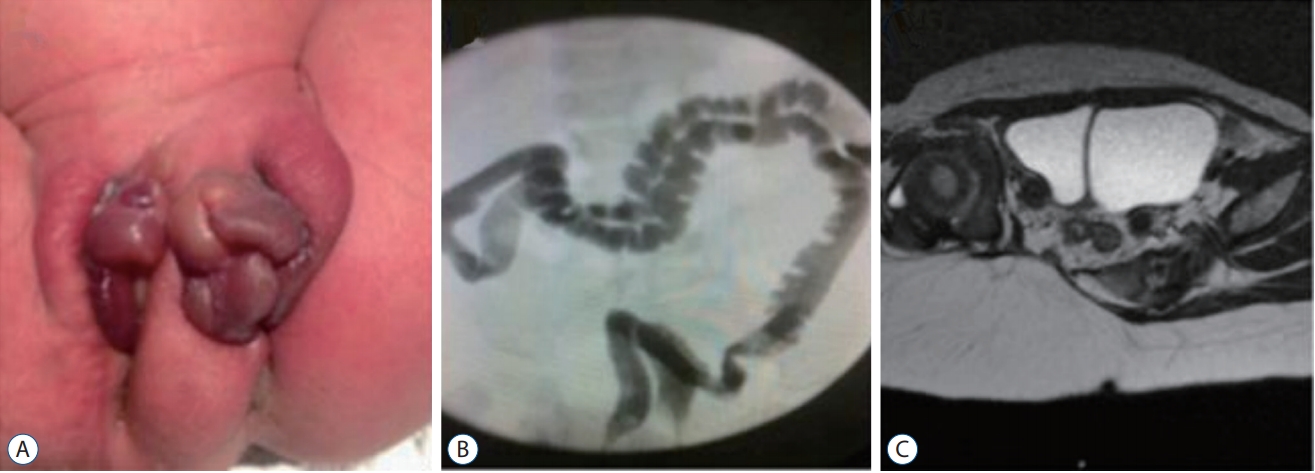
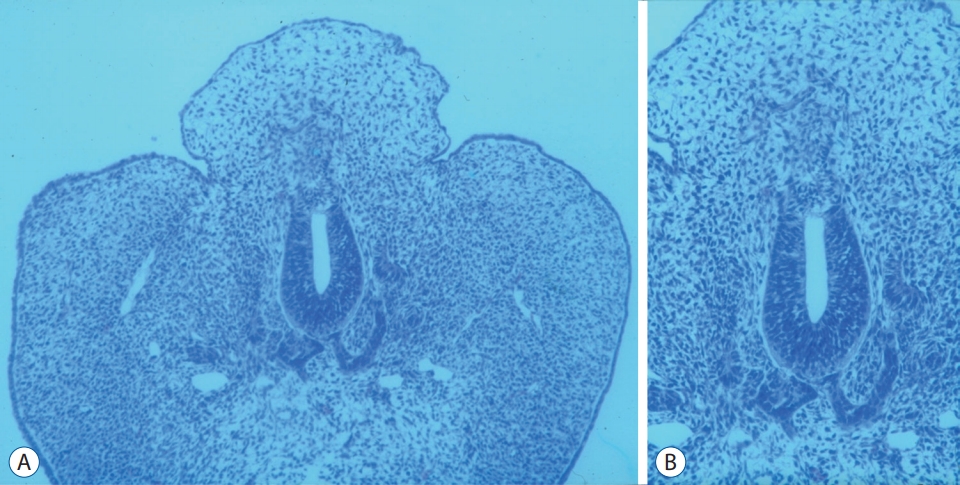
CDupS involves the gastrointestinal, genitourinary, spinal, musculoskeletal and cardiovascular systems. Among them, duplication from the transverse colon to the rectum, genitourinary organs and lower spine from the lower thoracic level are the most common. It is intriguing that the proximal portion of duplicated organs does not show a total independent duplication, meaning that the organs are separated by a septum but not as two independent sets of organs (Fig. 2). For example, duplicated colons run in parallel, sharing one septum between them. Similarly, a duplicated bladder or uterus shares a septum in the midline. From there, duplication occurs like the branching of a tree, followed by the formation of two sets of suborgans until reaching the distal end. Patients show two urethral, vaginal and anal orifices, although the bladder, uterus or rectum are only septated. The spine is duplicated from the lower thoracic region, branching off from one proximal spine. In contrast, a midline septum or neurenteric cyst between the two spinal cords, which are features of split cord malformation (SCM), are rare. Moreover, duplicated levels of CDupS are mostly in the lower spine, whereas SCMs do not have such a limitation. Apart from duplication, CDupS shows mesenchymal defects, which are also characteristics of caudal agenesis.
Mesenchymal defects such as gastroschisis, omphalocele, bladder exstrophy, and anorectal stenosis were reported in CDupS [1,3,15]. Interestingly, these findings were considered to be the results of caudal mesenchymal hypoplasia [33,46]. In fact, one interesting case with the coexistence of CDupS and caudal agenesis was reported [7]. Along with the common findings of CDupS, features shared with caudal agenesis also implicate a similar pathoembryogenesis in CDupS.
Regarding pathoembryogenesis theories, Dominguez et al. [15] speculated that CDupS originates from various insults that occur on the 23rd and 25th days of gestation, resulting in incomplete regression of Kovalevsky’s canal, also known as the neurenteric canal. The author postulated that the incomplete regression of the canal forms a fibrous band that divides the notochord and results in the duplication of the lower spine and spinal cord. The duplications of intestinal or genitourinary organs were explained as a result of the division of adjacent mesoderm and endoderm. The limitation of the proposed theory is that CDupS patients do not show vestiges of neurenteric components, such as neurenteric cysts or fistulas, which are frequently seen in SCM. In addition, the division of adjacent endoderm was insufficiently explained.
An approach to pathoembryogenesis from a different angle was attempted by Bannykh et al. [6] by focusing on ‘abnormal fusion’ rather than ‘abnormal duplication’. The authors proposed that CDupS may arise from partial fusion of an early embryo or two closely opposed individual notochords. For their theory to be possible, anomalous fusion of any part of the body axis, such as the cephalad or proximal trunk, should be seen. However, the noninvolved body portions have no evidence of anomalous fusion but are normal or almost normal. Furthermore, the septated condition of the intestines or bladders cannot be explained by this theory. The duplicated part, not the single part, is abnormal in CDupS. In addition, common manifestations of CDupS cases imply identical pathoembryogenetic spectrum with similar patterns in the similar organs, not a random event of fusion.
The two hemineural plates formed by the endomesenchymal tract that bisects the developing notochord due to adherence between the ectoderm and endoderm are a frequently quoted theory when reporting CDupS cases. However, the theory was used by Pang et al. [48] to explain the pathoembryogenesis of SCMs. As mentioned above, CDupS and SCM render two different entities. More importantly, the theory was applied to gastrulation and the primary neural tube but not to secondary neurulation and the secondary neural tube, which is an area of interest in CDupS [48].
Sur et al. [60] postulated that abnormal proliferation of the caudal mesenchyme may be led by a misexpression of distal Hox genes when explaining CDupS. However, the Hox gene is reported to be related to increased cell proliferation, showing overgrowth phenotypes in the tails of mutant mice, not a duplication [16]. We think that caudal mesenchymal hyperplasia may contribute to the abnormal overseptation of caudal structures. However, the associated clinical features of caudal agenesis, which are known to be caused by caudal mesenchymal hypoplasia, seem contradictory. Although the role of the Hox gene in CDupS could not be excluded, further experiments remain necessary to be clarified.
The important clue when inferring the pathoembryogenesis of CDupS through the common clinical findings is that the body axis is duplicated from one point. Duplicated organs are mostly derivatives of the hindgut and CCM. The body axis of an embryo is formed by the primitive streak and notochord, which is elongated rostrally from Hensen’s node. On postovulatory day 25-27, a primitive streak appears between the caudal neuropore and the cloacal membrane when the caudal neuropore closes [55]. From this point, the primitive streak is internalized and referred to as the CCM. In contrast to the most of the notochord, the caudal notochord arises from the CCM [42]. Therefore, the duplication of the caudal body axis may implicate the duplication of the CCM followed by the duplication of the caudal notochord.
The CCM is a primordium of a secondary neural tube, which is formed after the condensation, vacuolization and canalization of the cells [41]. The secondary neural tube gives rise to the terminal spinal cord, which encompasses the level of duplicated spinal cord shown in most cases of CDupS [41]. In addition, the hindgut, including the cloaca, gives rise to the transverse, descending, and sigmoid colon; the rectum and upper portion of the anal canal; and the bladder and urethra [39]. The formation of the hindgut begins at the fourth week of gestation from caudal folding of an embryo [39]. Hence, if CCM duplication occurs at the beginning of hindgut formation, which is located nearby, proximal organs derived from the hindgut would be affected by duplicated CCMs.
During hindgut formation, mesenchymal cells grow into the cloaca and divide it into the urogenital sinus and anorectal canal [70]. We speculated that parallel running of the CCM, or the late-stage primitive streak, would form mesenchymal cells between the two primordial body axes. As a result, the mesenchymal cells in between could form a septum between duplicated colons or bladders - the characteristic features of CDupS. The ectopic distribution of the mesenchyme has been shown to interfere with normal development [70]. Therefore, we propose that duplicated CCMs may form an intervening caudal midline mesenchyme, which later leads to aberrant insertion of mesenchymal tissue at the midline, which causes the septation of related organs (such as the bladder, uterus and colon) followed by the duplication of suborgans (such as the urethra, vagina and anus). On the other hand, the formation of intervening mesenchyme would also affect the mesenchyme placed bilateral to the two duplicated CCMs compared to the bilateral mesenchymal growth power in the case of only one CCM.
Ventral body wall defects such as gastroschisis, omphalocele or bladder exstrophy are caused by deficient ventral movement of lateral body folds, leading to incomplete fusion in the midline and rupture of the large cloacal membrane [53]. This process is accomplished by the end of the postovulatory 4th week. Hence, if the lateral mesenchyme lacks sufficient force for ventral push due to the leakage of the force to the intervening midline mesenchyme, ventral body wall defects may occur. Meanwhile, as stated above, abnormal midline mesenchymal movement may divide hindgut (including cloaca) derivatives, leading to CDupS (Fig. 3).
Another interesting finding of CDupS is the involvement of the heart. Because the heart develops from the rostral portion of the primitive streak [30,64], an insult during a certain stage of primitive streak development may also affect heart development.
Recent molecular biological findings are worth attention. Chordin, bone morphogenetic protein-4 (BMP-4), and GATA2 are reported to be involved in primitive streak formation. A double primitive streak and an ectopic primitive streak were formed by embryonic delivery of the Gata2 gene in chick embryos [8]. The misexpression of chordin also led to an ectopic primitive streak [59]. Primitive streak duplication occurred when the CYP26 gene, which is related to the degradation of retinoic acid, was deleted [61]. These data support our hypothesis.
CDupS is a rare congenital anomaly showing distal genitourinary, intestinal and spinal organ duplications. Although it remains elusive, the pathoembryogenesis of CDupS may be due to an insult during the late gastrulation phase by a molecular interaction, leading to ectopic primitive streak formation followed by CCM duplication. CCM duplication may subsequently cause the overactivity of abnormally positioned midline mesenchyme between the two CCMs and an underactive growth force of lateral and caudal mesenchyme. A more comprehensive study is mandatory to reveal the exact pathoembryogenesis of CDupS. Detailed explanations are available in our separate article [69].
FAILED INGRESSION OF THE PRIMITIVE STREAK TO THE CCM
ONTD (spina bifida aperta) has long been conceived to be the result of defects in only primary neurulation. The majority, if not all, of ONTDs consist of MMCs. The intuitive connection between the normal neurulation process of primary neurulation and the formation of skin and neural tube defects in MMCs has been undoubtedly accepted as the pathoembryogenetic mechanism. The normal process of secondary neural tube formation does not involve the rolling and fusion process of primary neurulation. Secondary neurulation consists of the condensation of the CCM and the cavitation of the medullary cord to form the secondary neural tube. Therefore, it is less intuitive to think that ONTDs can arise from errors during secondary neurulation [11].
However, MMCs in the region of secondary neurulation (below the S1-2 junction) are not uncommonly encountered. The skin defect and neural placode on the dorsal surface of the defect are identical to the MMC, so the only difference is the location of the lesion. It seems that low-lying MMCs are less likely to be associated with hydrocephalus or Chiari malformations than classic MMCs. Additionally, the rate of retethering is known to be higher for low-lying lesions, possibly due to the small caliber of the spinal canal [31]. An analysis of our own data revealed that these cases were not uncommonly found. We had cases with bony defects below the vertebral arch at the mid-sacral level and the distal spinal cords turning to the cranial direction (Fig. 4) and those with the distal spinal cords going straight to caudal skin defects (Fig. 5), as in the transitional and caudal types of LLM, respectively.
There may be two ways to explain ONTDs in the area of secondary neurulation. First, to be in line with the hypothesis that ONTDs can originate only from primary neurulation, one must think that the region of primary neurulation extends further down to the sacral level, which seems unlikely because some of the lesions are limited in the area of secondary neural tube.
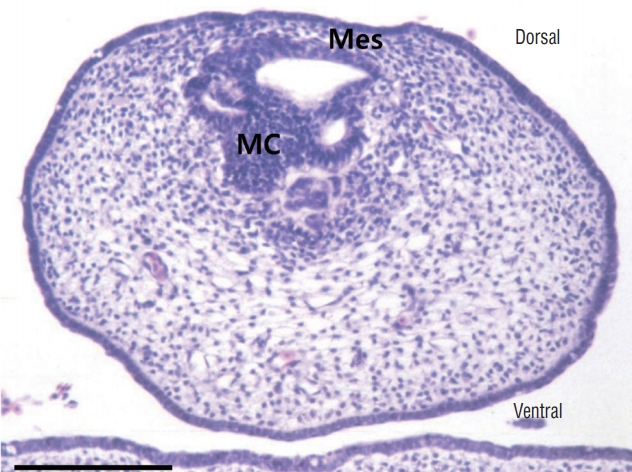
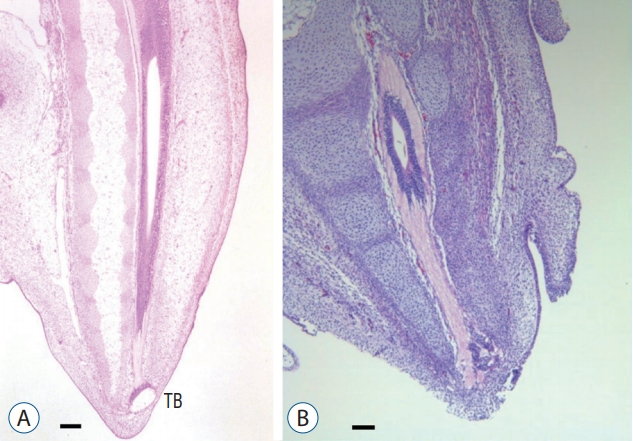
The next hypothesis would be that secondary neurulation error may also result in ONTDs. Studies on the development of neural tube development in chick embryos have provided clues to at least two possible mechanisms. 1) The CCM is a product of ingression from the primitive streak (Fig. 6). If ingression is hampered by some reason and there is a significant (not ‘focal limited’ as in limited dorsal myeloschisis [LDM]) continuation from the medullary cord to the remnant of the primitive streak that is exposed at the surface, an ONTD will form. In this case, the lesion may extend higher than the sacral hiatus, and the spinal cord may turn to the cranial direction. 2) The other is a well-known step during the regression phase of secondary neurulation, the ‘terminal balloon’ [32]. The terminal balloon is the dilated end of the degenerating medullary cord and has been found in chick embryo and human fetus specimens. A detailed description of the terminal balloon from formation to disappearance was performed using chick embryos. In some specimens, the dilatation was quite large and abutted the cutaneous ectoderm (Fig. 7). Another study by Schumacher [56] actually suggested that the rupture and subsequent healing of the terminal balloon can be observed as a part of the normal developmental process in chick embryos. We can postulate that the terminal balloon may rupture, leading to ONTDs in the region of secondary neurulation, either due to the nonhealing of a normally ruptured terminal balloon or the abnormal rupture of the terminal balloon. In this case, the lesion may be closely located at the sacral hiatus, and the spinal cord may go straight to the caudal skin defect. These two lesions may be compared with the transitional and caudal types of LLM, respectively (see ‘Neuro-mesenchymal adhesion’ below).
FOCAL LIMITED DORSAL NEURO-CUTANEOUS NONDISJUNCTION
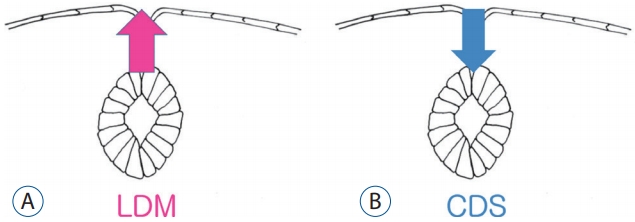
The clinical significance of congenital dermal sinus (CDS), a continuation of skin tissue into the deeper layers of the body, is well known : infection, mass effect by associated tumors and chemical inflammation by the leakage of the tumor contents. CDS is regarded as a product of failed normal disjunction of the primary neural tube from the cutaneous ectoderm, leaving a connecting stalk between them and drawing the skin tissue to the neural side. Inclusion tumors such as dermoid or epidermoid cysts may be formed at the stalk.
Many patients show similar findings to CDS on MRI but have different types of skin lesions: not a skin ostium but frequently a cigarette-burn scar. The squamous epithelial components are absent in the CDS-looking stalk. In 2010, Pang et al. [52] coined the term ‘LDM’ to these lesions and explained them as being due to the failure of normal disjunction of the neuroectoderm from the cutaneous ectoderm at the area of primary neurulation, pulling the neural tissue to the skin side, the direction opposite to that of CDS (Fig. 8). They classified LDMs into flat (noncystic) and cystic types.
It is not surprising that there are cases showing characteristics of both CDS and LDM. Considering the common pathoembryogenesis of CDS and LDM, all CDS, LDM and intermediate forms can be categorized in a single spectrum, ‘focal limited dorsal spinal neuro-cutaneous nondisjunction disorder’ [34,65].
The pathoembryogenesis of LDM shown in the article of Pang et al. [52] in 2010 can be attributed to an event occurring during primary neurulation, and LDM is believed to be an anomaly occurring only at the level of the primary neural tube.
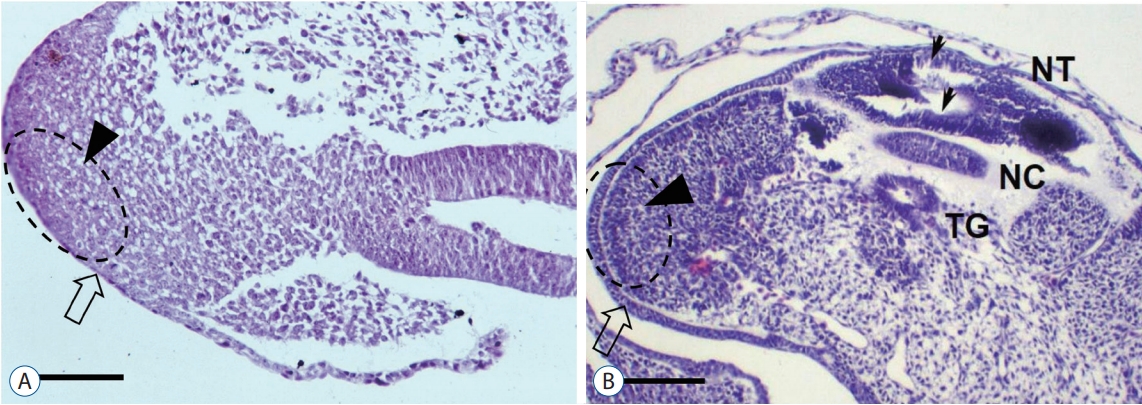
However, in addition to LDM located in the area of primary neural tube, we experienced low-lying cases of LDM. In the article of Pang et al. [52], the level of lesions was counted by the level of the vertebral body at the attachment of the LDM stalk to the dorsal spinal cord. We thought that this method of identifying the level of lesion is not reasonable because the spinal cord and spinal nerve root in the lower lumbar and sacral regions are located much higher than the corresponding level of the vertebral body. For example, S3 roots exit from the S3 segment of the spinal cord, a product of secondary neurulation. The exits from the spinal cord are located at the vertebral level of L1 or L2. However, the spinal roots traverse through the intervertebral foramen of the corresponding level, S3-4, before they reach the corresponding muscles and sensory apparatuses at the corresponding dermatome. Likewise, we postulated that a stalk connecting the skin area of a certain dermatome will pass through the interspinous ligament of the corresponding level before reaching the dorsal surface of the corresponding spinal cord segment (Fig. 9). Therefore, we counted the level of LDM lesions by the interspinous ligament level where the LDM stalk passes through.
It is known that the spinal cord below the S1-2 segment is formed by secondary neurulation, whereas the immediate cranial part up to the lower thoracic spinal cord is derived from the area of junctional neurulation [13,41]. In the area of junctional neurulation, the primary neural tube is located in the dorsal part. Therefore, LDM stalks passing through the interspinous ligament below S1-2 are attached to the spinal cord derived from the secondary neural tube, whereas LDM stalks passing through the interspinous ligament at S1-2 or higher may end at the primary neural tube.
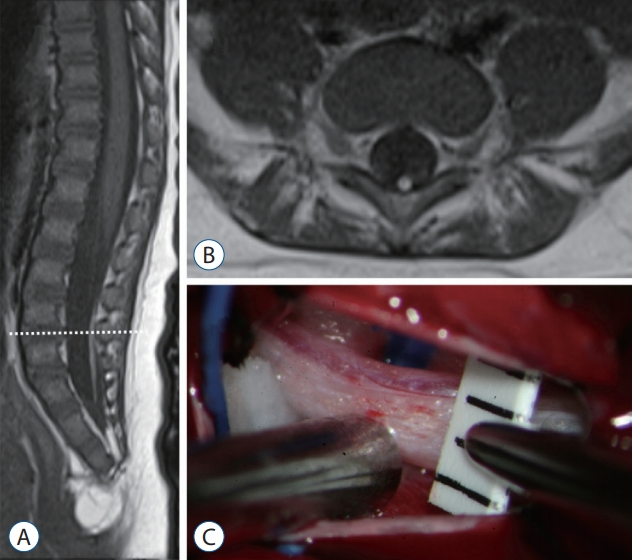
Recently, through the methods mentioned above, our team reported that LDM may be found in the area of secondary neural tube [27] (Fig. 10). In our series, approximately 40% (11/28) of all LDM cases were located in the area of secondary neural tube. Regarding the pathoembryogenesis of LDM, the continuation of the CCM with the surface-located primitive streak in the early phase of caudal body part formation explains how nondisjunction occurs (Fig. 6).
The counterpart of the same spectrum, CDS and its inclusion cysts, is also found in the area of secondary neural tube. Commonly, they are associated with other occult spinal dysraphisms, such as LLMs.
NEURO-MESENCHYMAL ADHESION
Since Chapman [10] morphologically classified LLMs at the conus (excluding filar type) into three types (dorsal, transitional and caudal) in 1982, the classification has been widely used. This classification is also useful for the planning of surgery, and there have been many efforts to correlate LLM type with pathoembryogenesis, clinical manifestations and management outcomes. Thereafter, modifications of the classification were proposed. They include the addition of lipomyelomeningoceles (an extraspinal extension with extraspinal herniation of the subarachnoid space) and a chaotic type (involving a placode and roots) [4,51].
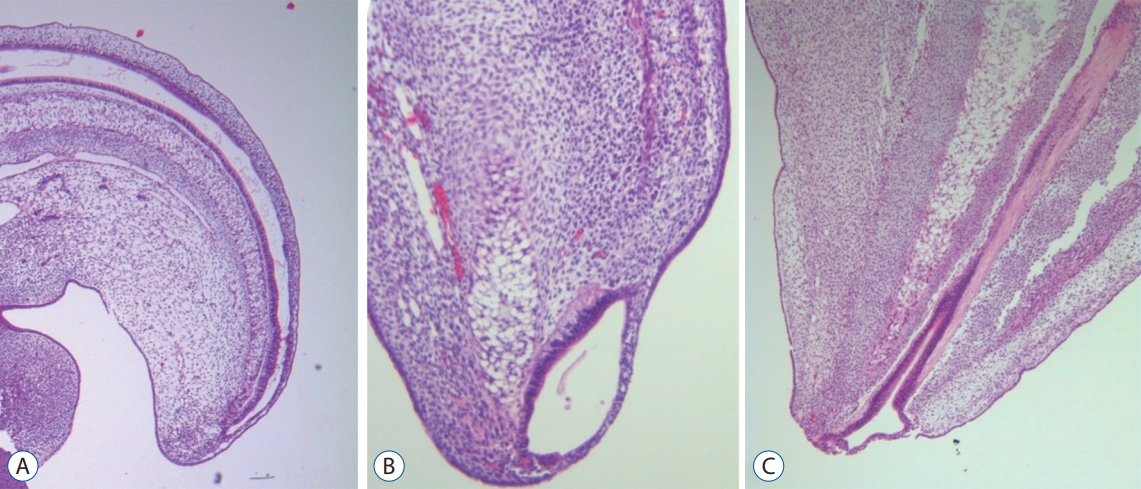
Regarding the pathoembryogenesis of dorsal-type LLM, the premature disjunction theory is widely accepted. According to McLone and Naidich [37], unilateral premature focal separation of the future neuroectoderm from the future cutaneous ectoderm at the neural fold provides a chance for intervening mesenchymal tissue to migrate into the developing neural tube, leading to dorsal myeloschisis with fat tissue filling the defect. The usual asymmetry of dorsal-type LLMs correlates well with the unilateral premature disjunction theory. Our team reported the results of experimental premature disjunction in chick embryos, supporting the premature disjunction theory [36]. Among the 35 embryos evaluated, one showed findings that suggested an LLM : a back lump with an acute angle at the junction with the surrounding normal skin, blending of the neuroepithelium and mesenchymal tissue through an indistinct basement membrane and notochordal abnormalities (Fig. 11).
Recently, Morota et al. [40] suggested a ‘new’ classification into four types based on embryonic stages. They included cases showing dorsal attachment of the fat mass down to the end of the conus with an identifiable interface between the conus and the fatty mass on MRI (previously classified as the transitional type) into the group of purely primary neurulation failure together with the previous dorsal type (type I). Type II denotes a transitional type that excludes those newly classified into type I (the failure of primary and secondary neurulations or the failure of junctional neurulation). Type III and type IV are the same as the previous caudal type (the failure of early secondary neurulation) and filar type (the failure of late secondary neurulation), respectively. We agree that their new classification has a high correlation with the presence and type of skin lesions, associated urogenital and anorectal anomalies, and the diversity of pathological components in the fatty mass. Additionally, it has high implications in surgical planning. However, we question whether lesions that involve the spinal cord down to the tip of the conus can be classified as ‘purely primary neurulation failure’ because the distal spinal cord below the junction of S1 and S2 spinal cord segments is known to be formed by secondary neurulation. In addition, they emphasized the timing of errors according to the LLM type, but the nature of errors was not mentioned for types II-IV.
The pathoembryogenesis of dorsal-type LLM is well known, but that of transitional, caudal and filar types has received less attention. We suggest a possible nature of errors in the pathoembryogenesis of transitional, caudal and filar types of LLM : ‘neuro-mesenchymal adhesion’ during secondary neurulation. Except for pure intradural lipomas (rare and prevalent at locations other than the lumbosacral area) and some LLMs of filar type, the main fat masses in LLMs are directly connected to subcutaneous fat. Occasionally, isolated small satellite fat masses are associated, but they are closely located to the main fat masses and seem to be chipped off from the main masses. The subcutaneous fat enters the spinal canal through the lamina defect (in dorsal and transitional types) or sacral hiatus (caudal type and some of filar type). This suggests neuromesenchymal adhesion rather than aberrant differentiation of hidden CCM chips to fat tissue as the pathoembryogenetic mechanism. If the multipotent cells in the CCM intermingle and remain in the medullary cord and differentiate into fat, the continuity to the subcutaneous fat in almost all cases of transitional and caudal types cannot be explained.
Transitional type
The CCM is separated from the primitive streak. Caudal structures, including the medullary cord, caudal notochord and caudal somites, originate from the CCM (Fig. 12). The primitive streak is the site of gastrulation. Epiblasts migrate through the primitive streak and change to mesodermal cells by epithelial-mesenchymal transition. Therefore, it is our speculation that there may be abnormal adhesion of the surface epithelium, CCM (separating from the primitive streak) and intervening mesenchymal tissue at the area of the dorsal midline. If the connection persists from the medullary cord (a product of the CCM) to the surface ectoderm (the site of the primitive streak), ONTDs or focal limited neuro-cutaneous nondisjunction disorders such as CDS or LDM may occur. If, however, the connection of the medullary cord persists only to the mesenchymal tissue, it may lead to LLM (Fig. 13). Considering that the caudal mesenchymal tissue adhered to the medullary cord originates from the multipotent cells of the CCM, it is not surprising that the fat tissue in this type of LLM may have various potential to be associated with mesenchymal components such as muscle, cartilage, bone, blood vessels, and even renal tissue [21,22] as well as dermoid cysts or peripheral nerve twigs from the adjacent skin and neural crest.
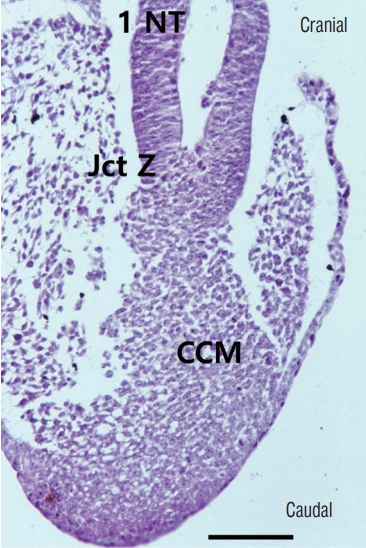
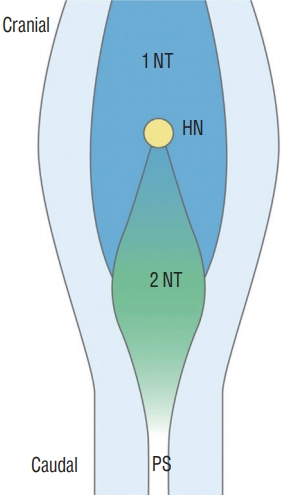
In the area of Hensen’s node, the future secondary neural tube is located at the caudal midline part of Hensen’s node, whereas the future primary neural tube is located at the lateral sides, and secondary neurulation progresses in the caudal direction [13,58] (Fig. 14). When neuro-mesenchymal adhesion occurs at the cranial part of the medullary cord, which is the caudal part of Hensen’s node, the closure of the caudal primary neural tube is also disturbed, and the dorsal neuro-adipose connection may extend up along the midline between the laterally located neural plates beyond the normal caudodorsal end of the primary neural tube (the junction of S1 and S2 spinal cord segments). The event may result in LLMs of the transitional type.
As mentioned above, if a transitional LLM is the result of differentiation error in the secondary neural tube, the fat tissue may be mainly present as an isolated lesion from the subcutaneous fat tissue and located at the ventral side of the spinal cord in the junctional zone (overlap zone between the primary and secondary neural tubes) because the secondary neural tube lies ventral to the primary neural tube in the junctional zone. However, in reality, the fat tissue in the junctional zone in transitional-type LLMs is located at the dorsal side and connects with the subcutaneous fat. This supports abnormal neuro-mesenchymal adhesion as the pathoembryogenetic mechanism rather than aberrant differentiation of hidden CCM chips to fat tissue in the secondary neural tube. In transitional-type LLMs, dorsal adhesion to mesenchymal tissue may involve the area down to the caudal end of the fully formed medullary cord. Wide exposure of the medullary cord to mesenchymal tissue allows an increased chance for double or multiple fat masses [40].
There may be minor modifications. 1) If the spinal cord and subarachnoid space are herniated through the lamina defect, a lipomyelomeningocele (an extraspinal extension type) is formed. Rarely, only the spinal cord is herniated out in the extraspinal extension type (lipomyelocele). 2) The medullary cord and adhered mesenchymal tissue may pull each other. Mostly, the mesenchymal tissue is pulled into the neural side, but occasionally, the spinal cord can be pulled out to the subcutaneous fat, especially in the extraspinal extension type where the traction force is higher by the protruding dome of the skin. 3) Frequently, the adhesion site is off the midline, and the developing laminae from both sides push the adhesion site to the midline rotating the spinal cord. 4) There is a chipping off of small mesenchymal fragments, especially when the connection between the medullary cord and mesenchymal tissue is thin, as shown in the filar type. These minor modifications may be applied to other types of LLMs, including the dorsal type.
Caudal type
If the abnormal neuro-mesenchymal adhesion site is pulled to the neural side at the stage when the upper medullary cord is well formed and only the distal end of the medullary cord is attached to the caudal mesenchymal tissue, a caudal-type LLM is formed (Figs. 15 and 16). The result is intraspinal extension of subcutaneous fat tissue through the sacral hiatus without the formation of the filum. As described in the ‘Regression failure spectrum of medullary cord’ section in this article, when abnormal neuro-mesenchymal adhesion occurs but there is no traction of mesenchymal tissue into the neural side, it may result in a retained medullary cord (RMC). If the spinal cord is distracted to the extraspinal side, a TMCC (if the terminal balloon persists) or a terminal myelocele (TMC, if the terminal balloon collapses) is formed.
Filar type
If the neuro-mesenchymal connection in the caudal type is thin and the involution of the distal medullary cord proceeds as normal (or close to normal), the result may be the filar-type LLM. When the fat tissue is slender, parts of the fat tissue may easily fall off from the main fat mass, resulting in satellite lesions.
If a focal neuro-mesenchymal connection occurs in the lower dorsal aspect (but not at the caudal end) of the medullary cord (similar to a minor form of the transitional type) and the involution of the distal medullary cord proceeds as normal, it may cause focal fat tissue in the filum. However, we think this is less likely because small focal fat masses in the filum never have connections to the dorsal subcutaneous fat tissue through the laminar defect or interspinous ligament.
REGRESSION FAILURE SPECTRUM OF THE MEDULLARY CORD
Secondary neural tube formation starts with condensation and proceeds sequentially with continued vacuolization, canalization, and regression. Anomalous lesions caused by errors in secondary neural tube formation can be largely divided into ‘failures of formation’ and ‘failures of regression’. Different entities are thought to arise depending on which stage the problem occurs during each process. All of the entities that we describe here are those that occur during the regression process, which is at the later stage of the secondary neural tube formation process. A failure of regression can result in the following entities: filar lesions, RMC, TMC, and TMCC.
Filar lesions
The filum is usually a thin and elastic structure that is a strand of tissue continuous from the tip of the conus medullaris to the coccyx in normal cases. However, patients with filar lesions have conditions such as a thickened filum terminale, fatty filum (filar lipoma) or filar cyst.
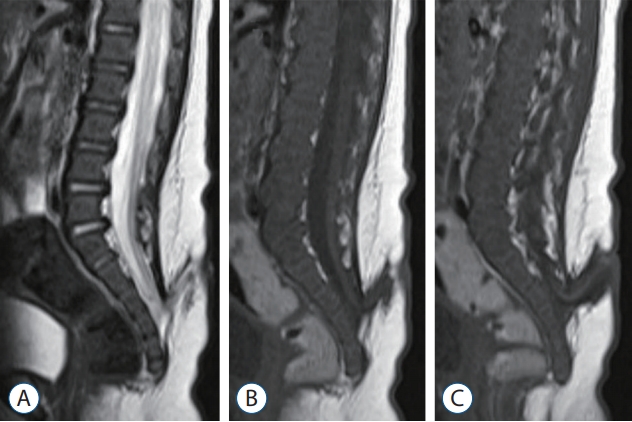
If fat or fibrous tissue grows into the filum and replaces glial tissue, it may create a fatty filum or thickened filum terminale (Fig. 17). The pathological filum (commonly defined as being more than 2 mm thick) may lose its elasticity and abnormally tether the spinal cord. Spinal cord traction can cause impaired microcirculation and oxidative metabolism of neural tissue, and ion channel dysfunction is directly related to neuronal membrane deformity (stretching) [66-68]. The patient can exhibit gait disturbance, pain, sensory deficit, urological dysfunction, scoliosis or foot deformities. Filar lesions are incidentally detected in 0.24-6% of the general population [2,9,18]. If symptoms exist, surgery is recommended, and prophylactic surgery can be performed before symptoms appear. Postoperatively, symptoms are improved or stabilized in 88% of patients, and retethering may occur in up to 5% [19,45].
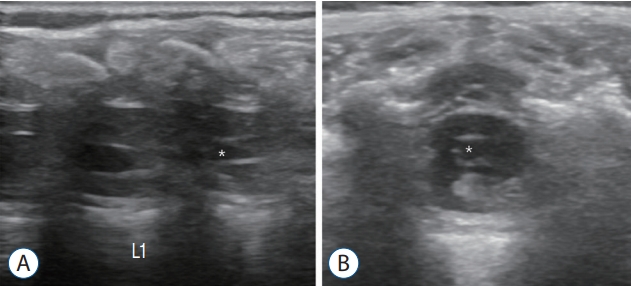
Some researchers believe that filar cysts (Fig. 18) are similar to the terminal ventricle and are caused by arrest during the regression process of the terminal ventricle. Filar cysts are usually asymptomatic and do not cause problems. Filar cysts may remain unchanged, shrink or even disappear on followup, which supports arrested or delayed regression. An isolated filar cyst is a benign entity and is not an indication for surgery [26].
RMC
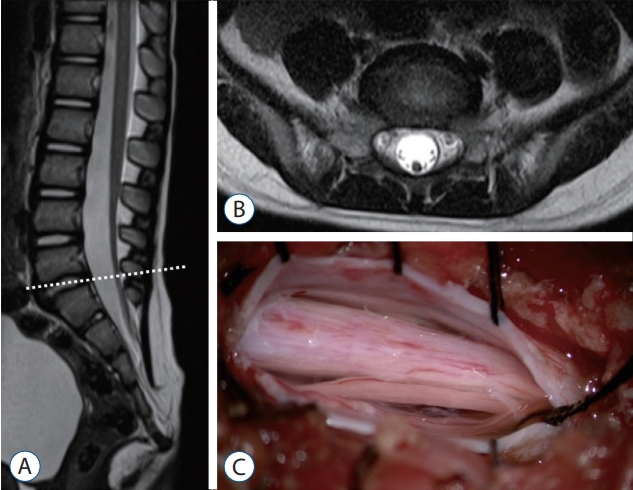
The term ‘RMC’ was first used by Pang et al. [50] to describe a spinal cord with a functionless segment continuous down to the cul-de-sac. It is assumed that the RMC is a medullary cord that remains as a result of regression failure in the process of secondary neurulation.
Conus medullaris descent from the normal position can be detected in RMCs through MRI. However, it is essential to confirm the diagnosis by checking for the presence of a caudal nonfunctional segment by IONM. An RMC shows no response to electrical stimulation.
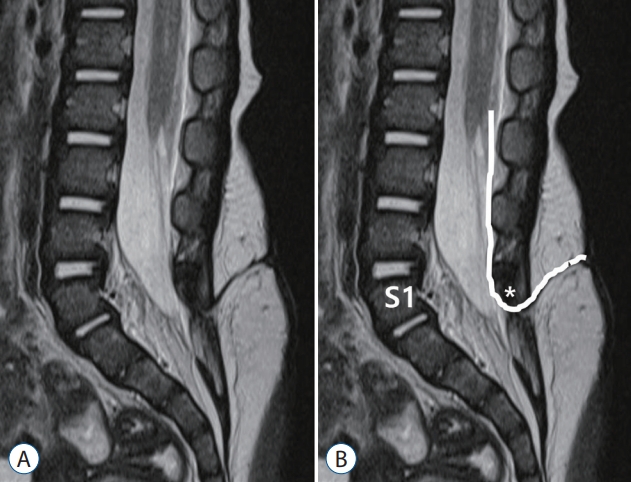
According to Pang et al. [50], the original definition of an RMC is a spinal cord that continues to the cul-de-sac without the formation of the thin filum terminale. However, Kim et al. [28] suggested that even if the tip of the conus is not connected to the cul-de-sac, if there is a portion of a nonfunctional distal cord, it could be an RMC. They defined an RMC as a spinal cord having a nonfunctional part in the distal conus (Fig. 19).
It is thought that some of the patients previously diagnosed with a low-lying conus may have nonfunctional parts and occasionally have cysts or fat masses. They may be regarded as having a ‘probable RMC’ if the region of question (distal conus) was not explored by IONM [28].
An RMC is believed to be caused by the arrest of regression during the process of secondary neurulation. However, there is no clear explanation for why arrest occurs. Many animal studies have suggested that ‘apoptosis’ is responsible for the regression of the medullary cord. Therefore, if ‘apoptosis’ does not occur at a time when it should occur, it can cause an RMC to form [23,25,38].
The clinical symptoms may include all the symptoms found in tethered cord syndrome, such as motor deficits, foot deformities, neurogenic bladder and bowel, frequent urinary tract infection, dysesthesia, leg pain, and coccydynia.
If the patient has symptoms, surgery is recommended, and sometimes, even if the patient does not have symptoms, surgery can be performed to prevent neurological events. During surgery, IONM must be performed. The surgeon must find the functionless medullary cord that does not respond to electrical stimulation, then cut the medullary cord safely and untether the RMC completely. Once safe and complete untethering is carried out using IONM, the prognosis of RMC seems good [47,54].
TMC
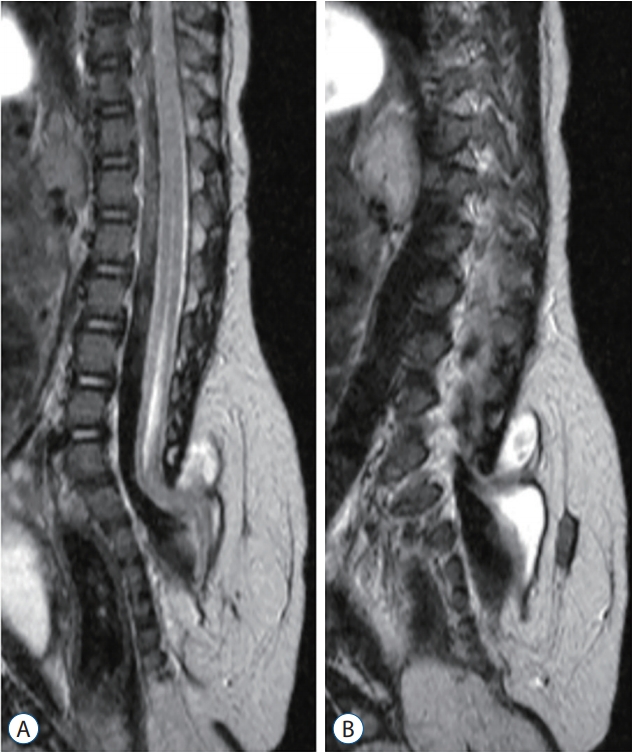
TMC is a more restrictive term than TMCC (described below). Similar to TMCC, TMC is a skin-covered spinal dysraphism in which the spinal cord is herniated along laminar and fascial defects. However, trumpet-like flaring, which is a terminal syringomyelic dilatation typically seen in TMCC, is not observed. Only an enlarged subarachnoid space and a herniated spinal cord and roots are responsible for the cystic contents (Fig. 20). This entity has the same pathoembryogenetical background with TMCC except the terminal balloon collapses. In fact, since the spinal cord is bound to subcutaneous fat tissue, it is reasonable to consider TMC as one of the extraspinal types of LLM.
TMCC
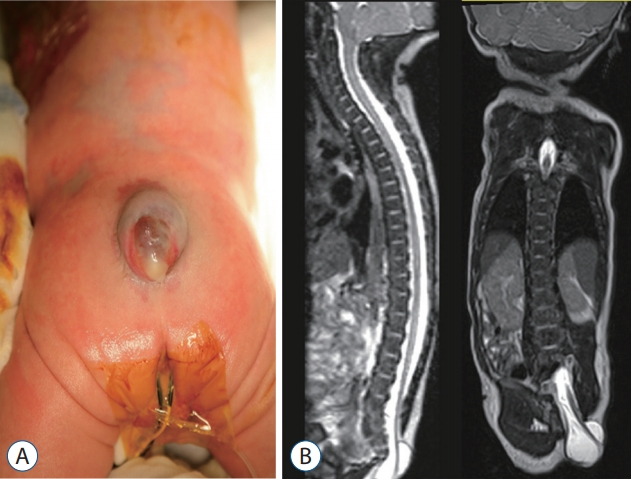
TMCC is the uncommon occurrence of spinal dysraphism in which the syringomyelic distal spinal cord and the enlarged subarachnoid space are herniated through the posterior lamina and fascia defect and covered by the skin. Similar to TMC, TMCC may be included in the extraspinal type of LLM.
TMCCs have essential features and nonessential features. As mentioned earlier, essential features include the caudal spinal cord that is stretched out of the spinal canal. The terminus of the spinal cord is adhered to the subcutaneous fat and appears to be shaped like a flared trumpet [49]. If the distal spinal cord does not have a syringomyelic cavity but other features are the same, it is a TMC, as described above.
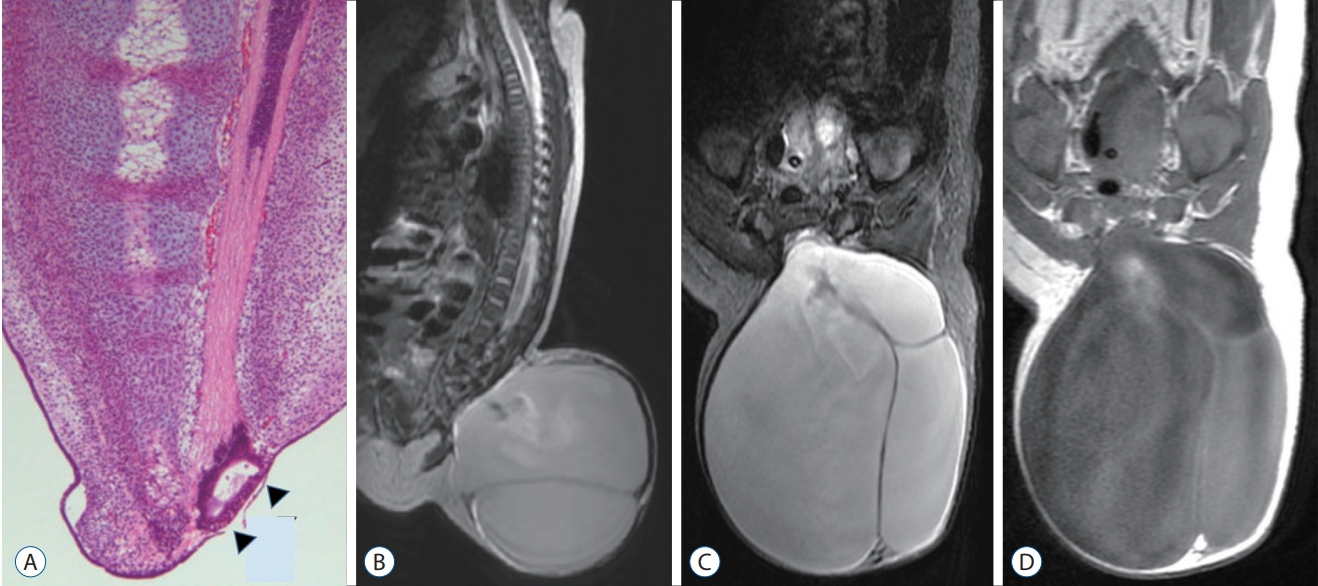
The pathoembryogenesis of TMCC is also considered a degeneration failure of secondary neurulation. Lee et al. [32] revealed the embryological background through research using chick embryos. During the secondary neurulation process, a ‘terminal balloon’ is developed at the caudal tip of the medullary cord. The distal end (dome) of the balloon is stuck to the skin of chick embryos. It had a shape like that of TMCCs (Fig. 21). Then it regresses as time passes, and the terminal balloon collapses and detaches from the skin. Schumacher [56] described the rupture and healing of the terminal balloon as a normal process in chick embryos. TMCCs are regarded as being the result of regression failure (arrest) at the phase of terminal balloon attachment to the skin.
TMCCs can be associated with other anomalies, such as an imperforate anus, ambiguous genitalia, omphalocele, bladder exstrophy and sacral bony anomalies. This is not surprising when TMC and TMCC are regarded as extraspinal variations of caudal-type LLM (type III by Morota et al. [40]) in which anorectal and urogenital anomalies may be associated. Hydrocephalus is also referred to as a related disease.
It is clear that a TMCC makes a tethered cord. However, the symptoms may not be noticed initially. Neurological deterioration may appear slowly as the child grows, but if the cyst increases rapidly, neurological deterioration can emerge suddenly by pulling the end of the spinal cord, which is attached to the dome of the cyst. As the cyst of TMCC increases rapidly, irreversible neurological deterioration may occur. Therefore early repair with resection is recommended. The cyst tends to enlarge in younger patients. The caution is needed in infancy [29,35]. For effective untethering and the prevention of retethering, IONM is essential for maximum elimination of the nonfunctioning cord during surgery. Details of the surgical procedure are beyond the scope of this article. With proper management, patients can have good-quality long-term survival.
CONCLUSION
Although connected to each other, secondary neurulation is rather an independent process from primary neurulation. Secondary neurulation has its own characteristics, such as relation to the skin and caudal mesenchyme or the caudal endoderm, a relatively absent neural-inductive role of the notochord, the presence of a regressive phase and the need for a junction with the primary neural tube. Disordered secondary neurulation at each phase of development may cause corresponding lesions, such as failed junction with the primary neural tube (JNTD, SSD), dysgenesis or duplication of the CCM associated with disturbed activity of caudal mesenchymal tissue (caudal agenesis and CDupS), failed ingression of the primitive streak to the CCM (MMC), focal limited dorsal neuro-cutaneous nondisjunction (LDMs and CDS), neuromesenchymal adhesion (LLM), regression failure spectrum of the medullary cord (thickened filum and filar cyst, RMC and low-lying conus, TMC, and TMCC). We found that almost every anomalous entity of primary neurulation can occur at secondary neurulation even though the pathoembryogenetical mechanisms are different. Furthermore, the close association with the activity of caudal mesenchymal tissue in secondary neurulation involves a wider range of surrounding structures than in primary neurulation. However, we do not exclude the possibility that multiple anomalies are caused by some molecular events or other primary events rather than by interactions among the structures involved as we suggested in this article.
Understanding secondary neurulation and its associated processes, such as gastrulation and junctional neurulation, may uncover the previously unknown pathoembryogenesis of the lesions that therefore have not been properly classified. Further revision of the classifications in spinal dysraphism is expected in the near future.