INTRODUCTION
Intracranial germ cell tumors (iGCTs) are a group of brain tumors with extraordinary characteristics. The cardinal features of brain tumors, such as age of onset, tumor location, histopathology and biological behavior, are quite distinct from other brain tumors of neuroepithelial origins. However, studies on molecular pathogenesis of iGCT lag behind the achievements noted for other brain tumors. The relatively low incidence and more importantly striking geographical difference of incidence precluded clinical interest and prevented global collaborative research for iGCT. Heterogeneity in patient ages and multiple pathological subgroups serve as a source of confusion between clinicians and researchers. Furthermore, the paradigm shift from radical surgery to biopsy with adjuvant therapies provided less tumor tissues for research than previously available. Despite these limitations, progress has been made in iGCT research, and our knowledge of the pathogenesis of iGCT has increased. In this review, we will present at overview current clinical knowledge on iGCT. Then, we will discuss the molecular pathogenesis of iGCT based on recent advancements.
EPIDEMIOLOGY OF IGCT
iGCT is histologically identical to GCT developing in other parts of the body. The majority of GCT cases arise from the gonads, i.e., testis and ovary. Therefore, a considerable amount of iGCT knowledge, such as histological classification, tumor markers, and even chemotherapeutic regimens, stemmed from the clinical experience of gonadal GCTs. iGCT is a special group of extragonadal GCTs that develops outside gonads. The most common sites of extragonadal GCTs include brain, mediastinum, retroperitoneum, and sacrococcyx [38]. Interestingly, all these sites are lined along the midline of the body, and many hypotheses have been proposed for this phenomenon. Even in the brain, iGCT develops in midline structures. Pineal and suprasellar regions are the most common sites for iGCT. Occasionally, iGCT is found at both sites simultaneously (so-called bifocal GCTs). Approximately 10% of iGCT arise from the basal ganglia, which is slightly off the midline. However, basal ganglia can be considered as midline structures separated by a narrow slit (the third ventricle). It is also noteworthy that all these locations are situated around the third ventricle. iGCT is rarely found in the cerebral hemisphere, cerebellum and spinal cord.
The striking geographical difference of iGCT incidence is interesting. iGCT is far more frequently diagnosed in East Asian countries, especially Korea and Japan. In the Korean Central Cancer Registry, the incidence of IGCT is 3.4/million/year [11]. The reported incidence is 2.7/million/year in Japan and 0.6/million/year in the USA [32]. In contrast, gonadal GCT, especially malignant testicular GCT, is much more common in Caucasians in Western countries (12/10000/year in Denmark, Norway, and Switzerland) compared with East Asia (2/10000/year in Japan) [56]. Malignant ovarian GCT occurs less frequently than testicular GCT, and no geographical difference is noted [56]. The difference in iGCT incidence according to sex is also interesting. In all countries, iGCT is more common in males. In Korea, the male-to-female incidence ratio is 4.53 : 1 [11]. In USA, the maleto-female ratio is 3.9 : 1 [29]. The male predominance of iGCT is more profound for pineal tumors compared with suprasellar tumors (13.0 : 1 and 1.73 : 1 in USA) [18]. Mediastinal GCT is also more common in males than in females, but sacrococcygeal GCT is more common in females [56].
Two age peaks are noted in iGCT incidence. A small peak exists for infants (0-2 years) and large peak stands for adolescents (13-19 years). Incidence declines rapidly after young adulthood. However, in a broader perspective including both gonadal and extragonadal GCTs, GCT appears to have three age peaks : infants, adolescents, and elderly. Infants mainly develop extragonadal GCTs, mostly in the brain and sacrococcyx. The majority of these infantile GCTs are teratoma (TE) and yolk sac tumor (YST) [38]. Infantile GCT is more common in females. In adolescents, both gonadal and extragonadal GCTs are increased. Seminoma (gonadal) and germinoma (GE) (extragonadal) are major pathologies. In elderly individuals, gonadal (testicular) GCT increases as a form of spermatocytic seminoma. Infantile brain TE/YSTs are typically large tumors situated in the third ventricle and are often diagnosed in utero with accompanying hydrocephalus. Extirpation of large brain tumors in the neonatal period presents serious challenges to surgeons and physicians. Infantile mature TE can recur as immature TE or YST when incompletely resected [39,60]. In adolescents, iGCT starts to develop around the onset of puberty. Therefore, hormonal influence is strongly suspected for iGCT development in adolescents and young adults.
HISTOPATHOLOGICAL SUBGROUP
As gonadal GCTs, iGCT is divided into two broad categories : GE and non-germinomatous GCT (NGGCT). NGGCT is further subdivided into four subgroups : TE, YST, choriocarcinoma (ChC), and embryonal carcinoma (EC). The distinction of GE from NGGCT is derived from the original theory of GCT pathogenesis, the so-called ŌĆśgerm cell theoryŌĆÖ proposed by Teilum [54]. In germ cell theory, GCT originates from primordial germ cells through neoplastic transformation. Each NGGCT arises from more differentiated stages of embryonic development starting from germ cells. Therefore, GE is a prototype of all GCTs. NGGCTs develop from more differentiated counterparts of embryonic and extraembryonic tissues.
The dualistic distinction of GE/NGGCT has more practical connotations than theoretical arguments. Although intracranial GE is a malignant tumor that spreads readily in the ventricles, GE is highly sensitive to radiation therapy (RT) and chemotherapy. GE is not considered a surgical disease. Therefore, biopsy followed by RT and chemotherapy is a standard protocol for GE. On the other hand, surgery plays a more important role for NGGCTs. Especially, TE is typically unresponsive to RT and chemotherapy, and surgery is the only therapeutic option for the disease. The prognosis of GE is excellent with >95% longterm survival. With the exception of benign mature TE (95-100% survival), NGGCT generally exhibits a poor prognosis compared with GE. Actually, the prognosis of YST and ChC was considered dismal until very recently. Therefore, from a clinical viewpoint, distinguishing GE from NGGCT is practical and highly recommended. In 1997, Dr. Matsutani of Japan proposed a scheme of prognosis groups of iGCTs, based on long-term treatment outcome [28]. GE and mature TE comprise the good prognosis group. Immature/malignant TE and mixed GCT consisting of GE/TE belong to the intermediate prognosis group. Highly malignant NGGCTs are included in the poor prognosis group. This scheme has been widely used in clinical practice. In Europe and North America, a dichotomous scheme of GE and NGGCT has been favored [31]. More specifically, division between tumor marker-positive and marker-negative iGCT is considered useful. In the European SIOP-CNS-GCT-96 trial, serum/cerebrospinal fluid (CSF) alpha fetoprotein (AFP) >1000 ng/mL was identified as a poor prognostic factor [6].
THE ENIGMA OF MIXED GCT
A confounding issue for iGCT subgrouping is the presence of mixed GCT. Mixed GCT has multiple components of individual iGCT subgroups. All mixed GCT logically belong to NGGCT. GE and TE are the most common components of mixed GCT. If malignant components, such as EC, YST, or ChC, are observed, the grade of the entire tumor is escalated to the poor prognosis group. Approximately 10-30% of iGCT are mixed GCT, but the proportion considerably increases if we search the entire paraffin block of pathological specimens for the trace of other components. If an iGCT mass is composed of 99% YST and 1% GE, can we call it mixed GCT or YST? If we miss the tiny 1% of GE in pathological diagnosis or the bit is not even included in surgical biopsy specimen, the diagnosis will be pure (100%) YST. It is not certain whether pure YST and 99% YST (+1% GE) are different diseases. However, small components in mixed GCT may be important in some instances. TE components in malignant mixed GCT can survive RT and chemotherapy. Paradoxical tumor growth is occasionally observed during or after adjuvant therapies. In this so-called ŌĆśgrowing teratoma syndromeŌĆÖ, mature or immature TE grows rapidly to an enormous size, causing a mass effect and hydrocephalus [25]. Early detection and surgical removal are critical for treatment.
DIAGNOSIS
Typical age of onset, sex, symptoms and image characteristics make the diagnosis straightforward in many instances. However, without clinical suspicion, it is occasionally very difficult to diagnose iGCT. Patients with suprasellar GCT typically present with diabetes insipidus that persists for months and even years. Loss of normal bright signal intensity in the posterior hypophysis is a characteristic finding [42]. Growth retardation and short statue are also common. If a tumor becomes large, visual disturbance and hydrocephalus can develop. Pineal GCT typically present with symptoms of hydrocephalus by obstruction of the cerebral aqueduct. Common symptoms and signs include headache, nausea, vomiting, visual disturbance, and abducens nerve palsy. ParinaudŌĆÖs syndrome with a classic triad of upward gaze palsy, light-near dissociation, and convergence-retraction nystagmus can be observed. Precocious puberty is occasionally found in patients with suprasellar or pineal GCT [42]. Beta human chorionic gonadotrophin (╬▓-HCG) secreted by GE and ChC components is associated with this phenomenon. Patients with basal ganglia GCT typically present with slowly progressive hemiparesis. Muscle atrophy and contracture are common but somatosensory function is preserved. The symptoms are so insidious that misdiagnosis is frequent. Atrophy of the ipsilateral pyramidal tract in medulla oblongata, cerebral peduncle, caudate nucleus, or cerebral hemisphere is a frequent finding on magnetic resonance imaging [40].
GE, especially of suprasellar origin, tends to be occult with invisible or very small lesion causing diabetes insipidus (socalled occult suprasellar GE) [23,30]. Basal ganglia GE sometimes exhibit similar insidious clinical course. The long latency between symptom onset and overt tumor progression of GE is an enigma and hormonal influence after puberty is suspected. Bifocal tumor involving both suprasellar and pineal areas constitute approximately 6-41% of iGCT [41]. Bifocal presentation has been regarded as a pathognomonic sign of GE, but some of the patients actually have mixed GCT [41]. It is not clear whether bifocal GCT is metastatic spread from one site or synchronous development. Ventricular seeding is common for GE, but diffuse leptomeningeal seeding over the cerebral cortex or spinal subarachnoid space is relatively uncommon. Therefore, the RT field should routinely include whole ventricles rather than limited to the tumor mass [31]. Craniospinal RT is indicated for patients with evidence of diffuse leptomeningeal seeding.
The presence of serum and CSF tumor markers is a unique characteristic of iGCT. ╬▓-HCG is markedly elevated in ChC, often greater than 10000 IU/L. ╬▓-HCG levels may be elevated in some patients with EC, immature teratoma, and GE. Increased serum/CSF ╬▓-HCG in GE is attributed to the presence of syncytiotrophoblasts but the possibility of mixed GCT should be considered. An arbitrary value of 50-100 IU/L is used to distinguish GE with syncytiotrophoblasts and mixed GCT with ChC components. The prognostic value of mild elevation of the ╬▓-HCG in GE is controversial. AFP is typically increased in YST, but its level is also elevated in some of EC and immature TE [43]. Usually, the ╬▓-HCG level is higher in CSF compared with serum, and the AFP level is increased in serum compared with CSF. Therefore, for the diagnosis of occult GE, assessment of CSF ╬▓-HCG is recommended [30]. However, tumor marker measurement methods are not standardized and cutoff values for diagnosis and risk stratification are not clearly defined.
TREATMENT
The treatment of iGCT should be multidisciplinary, incorporating surgery, chemotherapy, RT, and endocrine therapy. The major role of surgery is the acute management of hydrocephalus that frequently accompanies iGCT and tissue biopsy for diagnosis. With endoscopic procedures, neurosurgeons can achieve both goals in a single operation for suprasellar and pineal GCT. For basal ganglia GCT, a stereotactic biopsy is usually applied.
Treatment decision follows serum/CSF tumor marker expression. Marker-negative iGCT, that is, a tumor without elevated serum/CSF ╬▓-HCG and AFP, indicates GE or TE. Imaging can distinguish the two entities in many cases, but surgical biopsy is frequently undertaken to confirm the diagnosis. For mature TE, gross total resection provides disease cure. However, immature TE frequently requires adjuvant therapies [43]. Surgery has a limited role for GE except for biopsy and diagnosis. RT is the mainstay of treatment of GE. Previously, 36 Gy craniospinal RT with a tumor boost of 15 Gy was the standard treatment of GE, which yielded excellent outcome (>90% long-term survival) [46]. However, radiation can cause long-term sequelae of endocrinopathy, short statue, cognitive decline, and secondary malignancy. Therefore, RT fields and radiation doses have been reduced for GE. In a review of published data, whole brain or whole ventricular RT resulted in slightly more relapse (7.6%) than craniospinal RT with tumor boost (3.8%), but the difference was not statistically significant [46]. Focal RT should be avoided because it yielded an increased relapse rate (23.3%). Currently, localized GE is treated by whole brain or whole ventricular RT, with tumor doses of 36-39 Gy and whole ventricular doses of 19.5-24 Gy [8,9,35]. A short course of chemotherapy can be added before RT. The tumor outcome is excellent with >95% longterm survival. For disseminated GE, craniospinal RT is required.
Pre-radiation chemotherapy is commonly applied for GE. The aim of chemotherapy is not to enhance survival (survival rate is high enough with RT) but to reduce RT doses and potential complications of irradiation. The recently published SIOPCNS-GCT 96 trial demonstrated that chemotherapy followed by reduced volume and dose RT yielded comparable outcome with craniospinal RT alone [7]. Chemotherapy alone for GE has been attempted, but high rates of failure preclude this approach [24].
The prognosis of marker-positive iGCT is worse than that of GE and TE. Nearly all marker-positive iGCT are NGGCT except for GE with a mildly high level of ╬▓-HCG (<50-100 IU/L). Surgical resection, intensive chemotherapy, and craniospinal RT should be applied to treat malignant NGGCT. In 1990s, the reported 5-year-survival rate of malignant NGGCTs was approximately 30% [28,49]. Intensified multimodality treatment greatly enhanced the outcome of NGGCT. In recent retrospective studies on NGGCT, long-term survival rates of 74-80% were reported [26,27]. However, NGGCT is a very heterogeneous group of diseases with different prognoses. For example, mixed GCT mainly composed of GE plus immature TE definitely has better prognosis than pure YST or ChC. This heterogeneity makes comparison of outcomes from different studies, patient cohorts, and trials difficult. Recent trends show that the role of surgical resection for NGGCT is reduced. In the German MAKEI89 trial, surgery was not a significant factor for treatment outcome, but craniospinal RT and higher cisplatin dose were important [5]. Current standard for NGGCT is chemotherapy to shrink the tumor volume followed by craniospinal RT. The goal of chemotherapy for NGGCT is not to reduce radiation volume and dose, but to maximize the likelihood of survival [31]. During or after chemotherapy and RT, paradoxical growth of tumors (growing teratoma syndrome) should be treated by surgery. Second-look surgery is also considered for residual lesions after RT is complete. Published studies demonstrated that the majority of residual lesions were TE or scar tissues [36,58]. However, the indication and timing of second-look surgery are not established yet.
THEORIES ON CELLS OF ORIGIN FOR IGCT
There are diverse theories about the cells of origin for iGCT. These theories can be largely divided into two major streams often with minor modifications. The first one is traditional ŌĆśgerm cell theoryŌĆÖ. This theory dictates that gonadal GCT originates from primordial germ cells (PGC) through transformation. The development of extragonadal GCT is explained by the presence of ectopic germ cells that deviate from fetal PGC migration. In many animals, including humans, PGCs arising from the yolk sac epithelium are separated with somatic gonadal tissues and therefore should migrate to gonadal areas [45]. This migration process can give rise to ectopic germ cells in the midline of the body. GE is the neoplastic counterpart of PGCs or slightly more committed gonocytes. NGGCTs can develop from transformed PGCs/gonocytes through epigenetic reprogramming [38]. Thus, in germ cell theory, all iGCTs, including GE and NGGCT are truly germ cell tumors. The second theory is the so-called ŌĆśpluripotent stem cell theoryŌĆÖ. In this theory, seminoma/GE may originate from PGCs/gonocytes. NGGCT develops from embryonic stem (ES) cells with pluripotent potentials. EC is the prototype of all NGGCTs, which is the neoplastic counterpart of ES cells. YST, ChC, and TE can develop from the transformed EC cells. Extragonadal GCT can develop in a similar pattern from tissueresiding pluripotent stem cells through neoplastic transformation. The experimental fact that ES cells can give birth to germ cells leads to a suggestion that seminoma/GE may also develop from ES cells through the stage of PGCs/gonocytes. However, it cannot be settled by current evidence whether pluripotent stem cells are truly remnant ES cells (or ES-like cells) or reprogrammed PGCs.
SEMINOMA/GE ORIGINATES FROM GERM CELLS
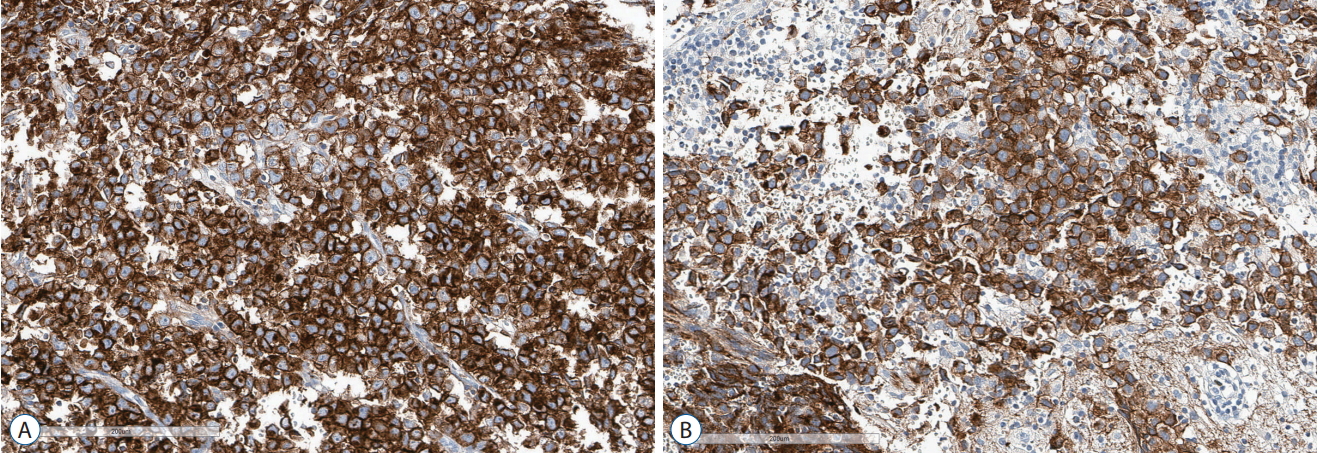
Germ cell origin of seminoma/GE is supported by multiple lines of evidence. First, the hypothesis was derived from the morphological resemblance of seminoma/GE cells to PGCs. The PGC is characterized by its large size and plump and round nucleus with conspicuous nucleolus, which are also typical features of seminoma/GE cells [12]. Second, seminoma/GE express pluripotency markers such as OCT4, NANOG, and SOX17, that are expressed by PGCs and ES cells [10,15]. Seminoma/GE also strongly express PLAP and KIT [33] (Fig. 1). PLAP and KIT represent markers of germ cell lineage differentiation derived from ES cells. Therefore, protein expression patterns support germ cell origin of the tumors. Third, PGCs undergo distinct changes of DNA methylation and demethylation during embryonic development. PGCs exhibit global erasure of methylation marks. PGCs gradually acquire methylation and sex-specific imprinting patterns during gametogenesis [21]. In testicular GCTs, seminoma exhibits global promotor hypomethylation with erasure of imprinting marks. The methylation status of seminoma closely resembles that of PGCs [34]. GE also exhibit similar global hypomethylation, supporting germ cell origin of seminoma/GE [14]. Fourth, PGCs are highly dependent on KIT signaling. KIT is a receptor tyrosine kinase and is crucial in the survival, proliferation, and migration of PGCs [50]. Its ligand, KITLG (also known as stem cell factor [SCF]) is provided by stromal cells of gonads [19]. KIT signaling is upstream of RAS-MAPK signaling and PI3K pathway, which are involved in a variety of cellular processes. Activating mutations in KIT and other genes in MAPK and PI3K pathways are the most common genomic variations found in both seminoma and GE [17,20]. KIT signaling is activated in seminoma/GE, reflecting PGCs as their cell of origin.
NGGCT MAY ORIGINATE FROM PLURIPOTENT STEM CELLS
As for gonadal and extragonadal NGGCT, the cell of origin problem is less clear than GE. In traditional germ cell theory, neoplastic germ cell (i.e., seminoma/GE) can evolve into more differentiated tissue-like tumors : YST resembling endodermal sinus, ChC similar to trophectoderm, and TE representing ectoderm, mesoderm and endoderm. However, the germ cell is not pluripotent. Germ cells need reprogramming to unchain its pluripotency potentials [38]. In the 1990s, Dr. Sano raised an intriguing question, namely, why more differentiated NGGCTs have worse prognosis than more primitive GE in TeilumŌĆÖs hierarchy [48]. Actually, cancers of undifferentiated histology occasionally exhibit better prognosis because RT and chemotherapy are more effective for rapidly proliferating, undifferentiated cells. Nonetheless, this query led to the suggestion that NGGCT may have different cells of origin from GE. The most plausible candidates are pluripotent stem cells residing in gonadal and extragonadal tissues.
In the 1970s, it was discovered that EC cells exhibit pluripotency. The research on EC cell pluripotency lead to the isolation of ES cells of mice in 1980s [3]. Thus, EC cells are regarded as neoplastic counterparts of ES cells. The robust expression of pluripotency markers, including OCT4, NANOG, and SOX2, in EC supports this conjecture [16]. Therefore, EC is considered to originate from neoplastic pluripotent stem cells. Then, more differentiated subtypes such as YST, ChC, and TE evolve from pluripotent stem cells with or without an intervening form of EC. Oosterhuis et al. [38] suggested dual cell of origins for NGGCTs. Infantile GCT, all of which are YST or TE, are close to ES cells, displaying partially erased imprinting marks, whereas post-pubertal NGGCT exhibit more completely erased imprinting patterns, resembling germ cells [38]. However, in testicular GCT, the genome of seminoma exhibits global hypomethylation, and all NGGCT subtypes have global hypermethylation similar to somatic tissues [34]. A recent large-scale methylation profiling of iGCT reveals that iGCT exhibits similar methylation patterns as testicular GCT : global hypomethylation in GE and hypermethylation in NGGCT [14]. If GCT retains methylation patterns after neoplastic transformation from the cell of origin, NGGCT does not seem to originate from PGCs/gonocytes but from more primitive stem cells-that are close to ES cells. The histological diversity of NGGCT can be attributed to the pluripotency of these stem cells.
THE GENOMIC LANDSCAPE OF IGCT
The importance of KIT signaling in germ cells led to interests in this proto-oncogene in testicular GCT. Earlier reports indicated high expression of c-KIT in seminoma but not in NGGCT. Activating KIT mutations are found in seminoma, especially in bilateral cases [4,55]. Studies have demonstrated that up to 25% of seminoma has mutations in KIT or KRAS genes, which are mutually exclusive. However, these mutations are rare in NGGCT [2,51]. In early studies before the era of next-generation sequencing, KIT mutation was also found in 25% of germinoma [22,47]. Interestingly, the most characteristic genetic event in testicular GCT is a gain of chromosome 12p, which is observed in almost all testicular GCTs. Isochromosome 12p [i(12p)] is the most frequent form (80%) and the remaining cases harbor duplication or focal amplification of 12p [44]. i(12p) is not present in infantile GCT and spermatocytic seminoma in elderly. Many genes on chromosome 12p have been implicated as driver oncogenes, i.e., KITLG, NANOG, KRAS, BCAT1, and CCND2, but none have been definitely proven [51]. At present, gain of chromosome 12p combined with activated KIT signaling appear to be the key molecular trigger in gonadal GCT pathogenesis.
However, gain of chromosome 12p material is less frequently observed in iGCTs. A study reported only 20% of iGCT had increased 12p including i(12p). The other study indicated that 25% of iGCT harbored i(12p) and 46% had polyploidy of chromosome 12 [37,52]. The discrepancy reflects small numbers of cases in each study, but it is clear that chromosome 12p gain plays a less crucial role in iGCT compared with testicular GCT. The other frequent chromosomal abnormality involve gain of X, 21q, and 14q. It is noteworthy that Kleinfelter syndrome (46, XXY) and Down syndrome (47, +21) are associated with extragonadal GCT including iGCT [1,53].
Recently, whole exome sequencing of iGCT tissues reveals mutations in KIT (26%), KRAS/NRAS (20%), CBL (11%), MTOR (8%), and NF1 (3%) [57]. KRAS/NRAS constitutes the downstream pathway of KIT receptor tyrosine kinase, feeding into MAPK pathways. Activating KIT and KRAS/NRAS mutations are mutually exclusive. CBL is a negative regulator of KIT-RAS signaling. NF1 is another negative regulator of RAS-MAPK pathway. A separate downstream pathway of KIT receptor consists of AKT1 and MTOR. Amplification of AKT1 is also observed in 19%. Overall, 53% of iGCT have one or more of genetic variations in the KIT-RAS-MAPK or AKT-MTOR pathways (Fig. 2). Interestingly, these genetic variations occur predominantly in GE. The same mutations are infrequently found in NGGCT. In another study on genomic features of iGCT, KIT mutation was found in 40% of GE and 6% of NGGCT [13]. RAS mutations were observed in 20% of GE and 3% of NGGCT. Many NGGCT cases with KIT/RAS mutations were actually mixed GCTs. Therefore, it is likely that, unlike GE, intracranial pure NGGCTs (i.e., pure EC, YST, ChC, and TE) are less dependent on KIT/RAS signaling.
An interesting study demonstrated that micro-dissected GE and NGGCT components of mixed GCT share the common KIT/RAS mutations, but differ in global methylation profile : hypomethylation in GE components and hypermethylation in NGGCT components [14]. Therefore, mixed GCT may develop from the same cell of origin, presumably PGCs with KIT/RAS mutation as an initiating event. Then, NGGCT components can be derived from GE through epigenetic reprogramming.
iGCT is characterized by peculiar geographic, age, and sex predilections. Genetic susceptibility has been suspected to explain the epidemiological features of iGCT. A rare germline variant of JMJD1C gene is enriched in iGCT patients, especially in Japanese patients [57]. The variant (S880P) is also enriched in the Japanese population. JMJD1C is a chromatin modifier and acts in germline development. More importantly, JMJD1C interacts with androgen receptor (AR) [59]. Through interaction with AR, the rare polymorphism of JMJD1C may account for the increased incidence in East Asia and male predominance of iGCT.
PERSPECTIVES
iGCT has attracted the interests of clinicians and researchers given the diverse histology, similarity to gonadal GCT, unusual epidemiological facts, and mysterious pathogenesis. From a clinical viewpoint, iGCTs are malignant brain tumors with the best prognosis currently known to neuro-oncologists. For GE, the treatment focus is shifting from survival to quality of life. RT dose reduction, endocrinological therapy, and psychosocial support are the main focuses of interest. For NGGCT, more promising outcomes are being unleashed with multimodal therapies. Actually, iGCT can be an index disease where surgery, chemotherapy, and RT all contribute greatly and effectively to enhance patientsŌĆÖ outcomes. Recent advancements in genomewide analysis reveal very interesting findings regarding underlying genetic mutations, altered signaling, and most importantly methylation profiling. This research can lead us not only to a new hypothesis of iGCT pathogenesis but also to novel therapy targeting the aberrant signaling pathways. KIT mutations are commonly found in chronic myeloid leukemia and gastrointestinal stromal tumors. Tyrosine kinase inhibitors, such as imatinib mesylate and dasatinib showed fair efficacy against these KIT-activated human malignancies. Although the prognosis of GE is excellent by current protocols, some patients develop recurrence. These patients can be salvaged by tyrosine kinase inhibitors in the future. Furthermore, a novel therapy is highly requested for NGGCT, and more studies are needed to define the therapeutic targets in NGGCT.